

Translating Lupus: Comparative Transcriptional Profiles of Preclinical Lupus Models and Their Relevance to Human Disease

 and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Husbandry
2.2. Accelerated Mouse Models of SLE
2.2.1. Poly (I:C)-Induced Lupus Nephritis Model
2.2.2. Mouse IFNα (mIFNα)-Induced Lupus Nephritis Model
2.2.3. Bm12 Lupus Nephritis Model
2.3. Spontaneous Mouse Models of SLE
2.3.1. NZB/W Spontaneous Lupus Nephritis Model
2.3.2. MRL/Lpr Lupus Model
2.4. Anti-Double-Stranded DNA (dsDNA) Assay
2.5. Quantification of RNA from Mouse Tissue
2.6. Statistical Analysis
2.6.1. Per-Marker Analysis in Animal Models
2.6.2. Pathway Analysis of Differential Expression in Animal Models
2.6.3. Pathway Analysis of Association of Animal Models to Human Bulk RNA Sequencing and to Human scRNA Sequencing (CD4, CD8, B, NK)
3. Results
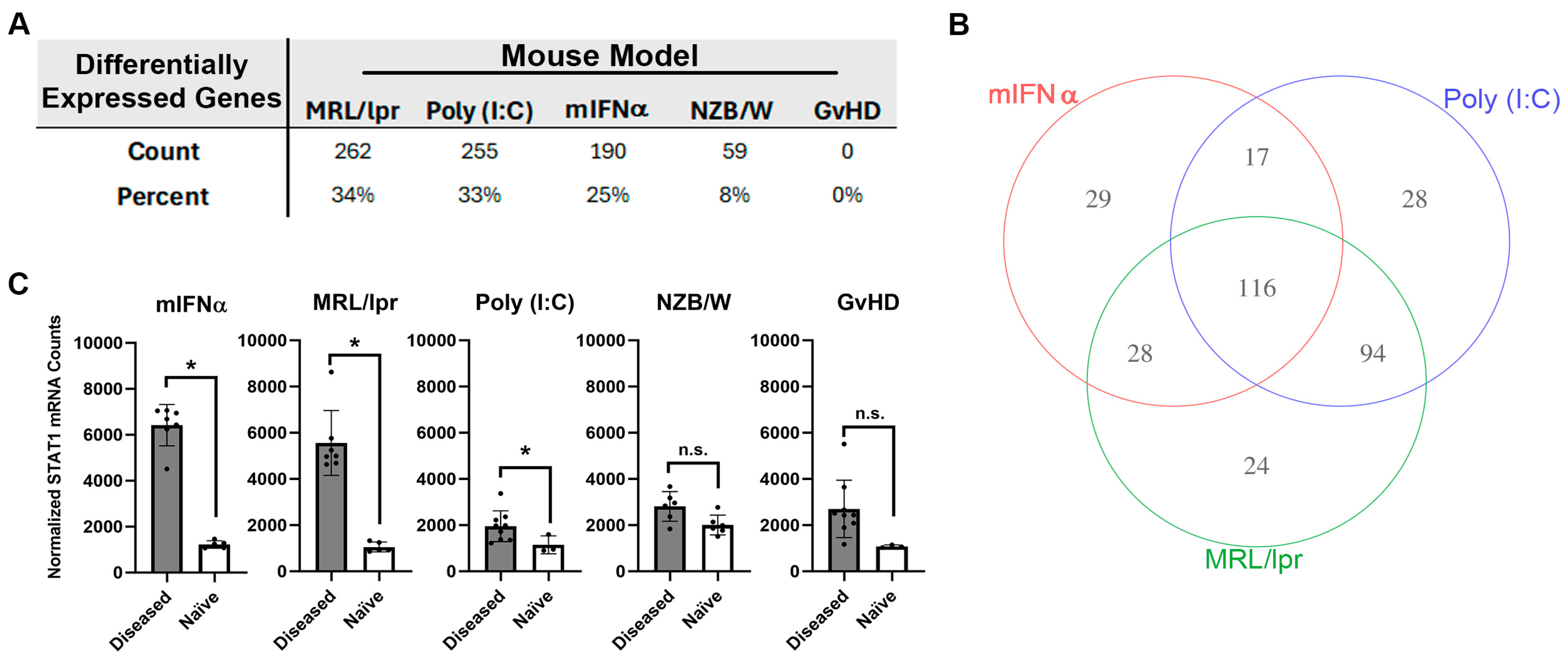
3.1. Differential Gene Expression in Kidney Tissue from Animal Models of SLE/LN
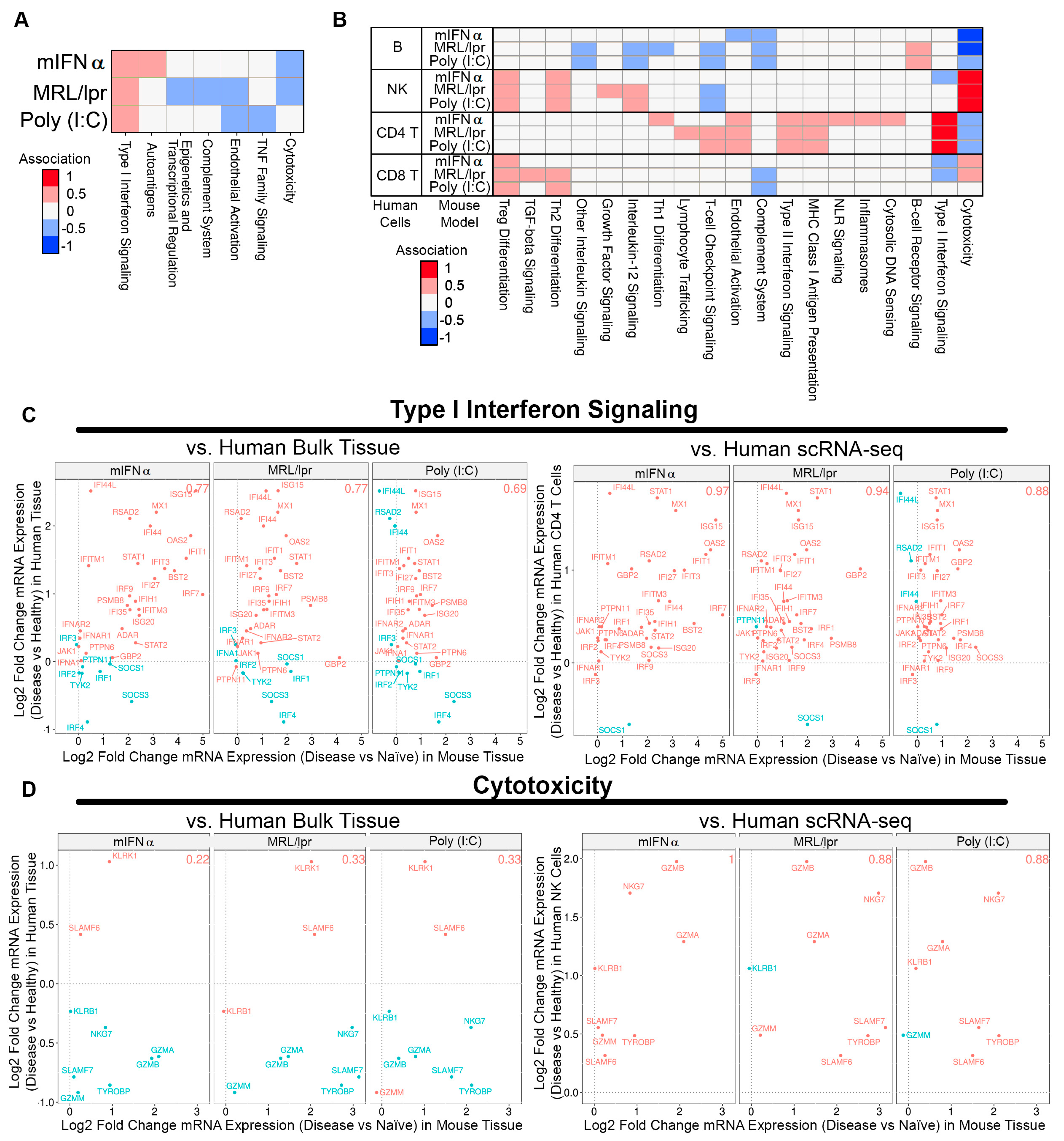
3.2. Comparative Pathway Analysis between SLE/LN Animal Models
3.3. Association between Animal Tissue and Human Bulk Tissue or Single-Cell Gene Signatures in SLE Kidney
4. Discussion
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bertsias, G.K.; Salmon, J.E.; Boumpas, D.T. Therapeutic opportunities in systemic lupus erythematosus: State of the art and prospects for the new decade. Ann. Rheum. Dis. 2010, 69, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Fanouriakis, A.; Tziolos, N.; Bertsias, G.; Boumpas, D.T. Update οn the diagnosis and management of systemic lupus erythematosus. Ann. Rheum. Dis. 2021, 80, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Hoover, P.J.; Costenbader, K.H. Insights into the epidemiology and management of lupus nephritis from the US rheumatologist’s perspective. Kidney Int. 2016, 90, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Bajema, I.M.; Wilhelmus, S.; Alpers, C.E.; Bruijn, J.A.; Colvin, R.B.; Cook, H.T.; D’Agati, V.D.; Ferrario, F.; Haas, M.; Jennette, J.C.; et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: Clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 2018, 93, 789–796. [Google Scholar] [CrossRef]
- Arazi, A.; Rao, D.A.; Berthier, C.C.; Davidson, A.; Liu, Y.; Hoover, P.J.; Chicoine, A.; Eisenhaure, T.M.; Jonsson, A.H.; Li, S.; et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 2019, 20, 902–914. [Google Scholar] [CrossRef]
- Dubois, E.L.; Horowitz, R.E.; Demopoulos, H.B.; Teplitz, R. NZB/NZW mice as a model of systemic lupus erythematosus. JAMA 1966, 195, 285–289. [Google Scholar] [CrossRef]
- Liang, B.; Gardner, D.B.; Griswold, D.E.; Bugelski, P.J.; Song, X.Y. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology 2006, 119, 296–305. [Google Scholar] [CrossRef]
- Mina-Osorio, P.; LaStant, J.; Keirstead, N.; Whittard, T.; Ayala, J.; Stefanova, S.; Garrido, R.; Dimaano, N.; Hilton, H.; Giron, M.; et al. Suppression of glomerulonephritis in lupus-prone NZB × NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 2013, 65, 2380–2391. [Google Scholar] [CrossRef]
- Rankin, A.L.; Seth, N.; Keegan, S.; Andreyeva, T.; Cook, T.A.; Edmonds, J.; Mathialagan, N.; Benson, M.J.; Syed, J.; Zhan, Y.; et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J. Immunol. 2013, 191, 4540–4550. [Google Scholar] [CrossRef]
- Haas, K.M.; Watanabe, R.; Matsushita, T.; Nakashima, H.; Ishiura, N.; Okochi, H.; Fujimoto, M.; Tedder, T.F. Protective and pathogenic roles for B cells during systemic autoimmunity in NZB/W F1 mice. J. Immunol. 2010, 184, 4789–4800. [Google Scholar] [CrossRef]
- Boneparth, A.; Woods, M.; Huang, W.; Akerman, M.; Lesser, M.; Davidson, A. The Effect of BAFF Inhibition on Autoreactive B-Cell Selection in Murine Systemic Lupus Erythematosus. Mol. Med. 2016, 22, 173–182. [Google Scholar] [CrossRef]
- Sharabi, A.; Haviv, A.; Zinger, H.; Dayan, M.; Mozes, E. Amelioration of murine lupus by a peptide, based on the complementarity determining region-1 of an autoantibody as compared to dexamethasone: Different effects on cytokines and apoptosis. Clin. Immunol. 2006, 119, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhu, T.; Bastacky, S.; McHale, T.; Li, J.; Xiao, X. Prevention and reversal of lupus in NZB/NZW mice by costimulatory blockade with adeno-associated virus-mediated gene transfer. Arthritis Rheum. 2005, 52, 3975–3986. [Google Scholar] [CrossRef] [PubMed]
- Mathian, A.; Weinberg, A.; Gallegos, M.; Banchereau, J.; Koutouzov, S. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J. Immunol. 2005, 174, 2499–2506. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bethunaickan, R.; Huang, W.; Lodhi, U.; Solano, I.; Madaio, M.P.; Davidson, A. Interferon-α accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 2011, 63, 219–229. [Google Scholar] [CrossRef]
- Triantafyllopoulou, A.; Franzke, C.W.; Seshan, S.V.; Perino, G.; Kalliolias, G.D.; Ramanujam, M.; van Rooijen, N.; Davidson, A.; Ivashkiv, L.B. Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc. Natl. Acad. Sci. USA 2010, 107, 3012–3017. [Google Scholar] [CrossRef]
- Steinberg, A.D.; Baron, S.; Talal, N. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc. Natl. Acad. Sci. USA 1969, 63, 1102–1107. [Google Scholar] [CrossRef]
- Liu, Z.; Bethunaickan, R.; Huang, W.; Ramanujam, M.; Madaio, M.P.; Davidson, A. IFN-α confers resistance of systemic lupus erythematosus nephritis to therapy in NZB/W F1 mice. J. Immunol. 2011, 187, 1506–1513. [Google Scholar] [CrossRef]
- Watson, M.L.; Rao, J.K.; Gilkeson, G.S.; Ruiz, P.; Eicher, E.M.; Pisetsky, D.S.; Matsuzawa, A.; Rochelle, J.M.; Seldin, M.F. Genetic analysis of MRL-lpr mice: Relationship of the Fas apoptosis gene to disease manifestations and renal disease-modifying loci. J. Exp. Med. 1992, 176, 1645–1656. [Google Scholar] [CrossRef]
- Andrews, B.S.; Eisenberg, R.A.; Theofilopoulos, A.N.; Izui, S.; Wilson, C.B.; McConahey, P.J.; Murphy, E.D.; Roths, J.B.; Dixon, F.J. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 1978, 148, 1198–1215. [Google Scholar] [CrossRef]
- Watanabe, H.; Kubo, M.; Taniguchi, A.; Asano, Y.; Hiramatsu-Asano, S.; Ohashi, K.; Zeggar, S.; Katsuyama, E.; Katsuyama, T.; Sunahori-Watanabe, K.; et al. Amelioration of nephritis in receptor for advanced glycation end-products (RAGE)-deficient lupus-prone mice through neutrophil extracellular traps. Clin. Immunol. 2023, 250, 109317. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Hermann, F.J.; Schmidbauer, K.; Talke, Y.; Rodriguez Gomez, M.; Schiechl, G.; Schlossmann, J.; Brühl, H.; Anders, H.J.; Mack, M. IL-3 contributes to development of lupus nephritis in MRL/lpr mice. Kidney Int. 2015, 88, 1088–1098. [Google Scholar] [CrossRef]
- Morris, S.C.; Cohen, P.L.; Eisenberg, R.A. Experimental induction of systemic lupus erythematosus by recognition of foreign Ia. Clin. Immunol. Immunopathol. 1990, 57, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Maldonado, M.A.; Madaio, M.; Eisenberg, R.A. The role of host (endogenous) T cells in chronic graft-versus-host autoimmune disease. J. Immunol. 1998, 161, 5880–5885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.D.; Gant, T.W. A simple and robust method for connecting small-molecule drugs using gene-expression signatures. BMC Bioinform. 2008, 9, 258. [Google Scholar] [CrossRef]
- Yao, M.; Gao, C.; Zhang, C.; Di, X.; Liang, W.; Sun, W.; Wang, Q.; Zheng, Z. Identification of Molecular Markers Associated With the Pathophysiology and Treatment of Lupus Nephritis Based on Integrated Transcriptome Analysis. Front. Genet. 2020, 11, 583629. [Google Scholar] [CrossRef]
- Aue, A.; Szelinski, F.; Weißenberg, S.Y.; Wiedemann, A.; Rose, T.; Lino, A.C.; Dörner, T. Elevated STAT1 expression but not phosphorylation in lupus B cells correlates with disease activity and increased plasmablast susceptibility. Rheumatology 2020, 59, 3435–3442. [Google Scholar] [CrossRef]
- NIAMS. Accelerating Medicines Partnership® Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP® RA/SLE) Program. Available online: https://www.niams.nih.gov/grants-funding/funded-research/accelerating-medicines/RA-SLE (accessed on 1 September 2020).
- Mahieu, M.A.; Strand, V.; Simon, L.S.; Lipsky, P.E.; Ramsey-Goldman, R. A critical review of clinical trials in systemic lupus erythematosus. Lupus 2016, 25, 1122–1140. [Google Scholar] [CrossRef]
- Pawar, R.D.; Patole, P.S.; Ellwart, A.; Lech, M.; Segerer, S.; Schlondorff, D.; Anders, H.J. Ligands to nucleic acid-specific toll-like receptors and the onset of lupus nephritis. J. Am. Soc. Nephrol. 2006, 17, 3365–3373. [Google Scholar] [CrossRef]
- Adalid-Peralta, L.; Mathian, A.; Tran, T.; Delbos, L.; Durand-Gasselin, I.; Berrebi, D.; Peuchmaur, M.; Couderc, J.; Emilie, D.; Koutouzov, S. Leukocytes and the kidney contribute to interstitial inflammation in lupus nephritis. Kidney Int. 2008, 73, 172–180. [Google Scholar] [CrossRef]
- Liao, X.; Ren, J.; Reihl, A.; Pirapakaran, T.; Sreekumar, B.; Cecere, T.E.; Reilly, C.M.; Luo, X.M. Renal-infiltrating CD11c(+) cells are pathogenic in murine lupus nephritis through promoting CD4(+) T cell responses. Clin. Exp. Immunol. 2017, 190, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Cabana-Puig, X.; Bond, J.M.; Wang, Z.; Dai, R.; Lu, R.; Lin, A.; Oakes, V.; Rizzo, A.; Swartwout, B.; Abdelhamid, L.; et al. Phenotypic Drift in Lupus-Prone MRL/lpr Mice: Potential Roles of MicroRNAs and Gut Microbiota. Immunohorizons 2022, 6, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.M.; Edwards, M.R.; Mu, Q.; Yu, Y.; Vieson, M.D.; Reilly, C.M.; Ahmed, S.A.; Bankole, A.A. Gut Microbiota in Human Systemic Lupus Erythematosus and a Mouse Model of Lupus. Appl. Environ. Microbiol. 2018, 84, e02288-17. [Google Scholar] [CrossRef] [PubMed]
- Cansu, D.; Temiz, G.; Açıkalın, M.F.; Korkmaz, C. Pauci-immune lupus nephritis: Possibility or co-incidence? Eur. J. Rheumatol. 2017, 4, 73–75. [Google Scholar] [CrossRef]
- Akhtar, M.; al-Dalaan, A.; el-Ramahi, K.M. Pauci-immune necrotizing lupus nephritis: Report of two cases. Am. J. Kidney Dis. 1994, 23, 320–325. [Google Scholar] [CrossRef]
- Richard, M.L.; Gilkeson, G. Mouse models of lupus: What they tell us and what they don’t. Lupus Sci. Med. 2018, 5, e000199. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, J.T.; Chang, C.-Y.; Kersjes, K.; Higgins, I.A.; Vendel, A.C.; Chang, W.Y. Translating Lupus: Comparative Transcriptional Profiles of Preclinical Lupus Models and Their Relevance to Human Disease. Biology 2024, 13, 778. https://doi.org/10.3390/biology13100778
Parker JT, Chang C-Y, Kersjes K, Higgins IA, Vendel AC, Chang WY. Translating Lupus: Comparative Transcriptional Profiles of Preclinical Lupus Models and Their Relevance to Human Disease. Biology. 2024; 13(10):778. https://doi.org/10.3390/biology13100778
Chicago/Turabian StyleParker, James T., Ching-Yun Chang, Kara Kersjes, Ixavier A. Higgins, Andrew C. Vendel, and William Y. Chang. 2024. "Translating Lupus: Comparative Transcriptional Profiles of Preclinical Lupus Models and Their Relevance to Human Disease" Biology 13, no. 10: 778. https://doi.org/10.3390/biology13100778