
GBMPhos: A Gating Mechanism and Bi-GRU-Based Method for Identifying Phosphorylation Sites of SARS-CoV-2 Infection


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. One-Hot Encoding
2.2.2. BLOSUM62
2.2.3. ZScale
2.2.4. Binary_5bit_Type 1
2.2.5. Binary_5bit_Type 2
2.2.6. CNN
2.2.7. Bi-GRU
2.2.8. Fully Connected Layer
2.3. Performance Evaluation
3. Results
3.1. Optimizing of Different Window Sizes
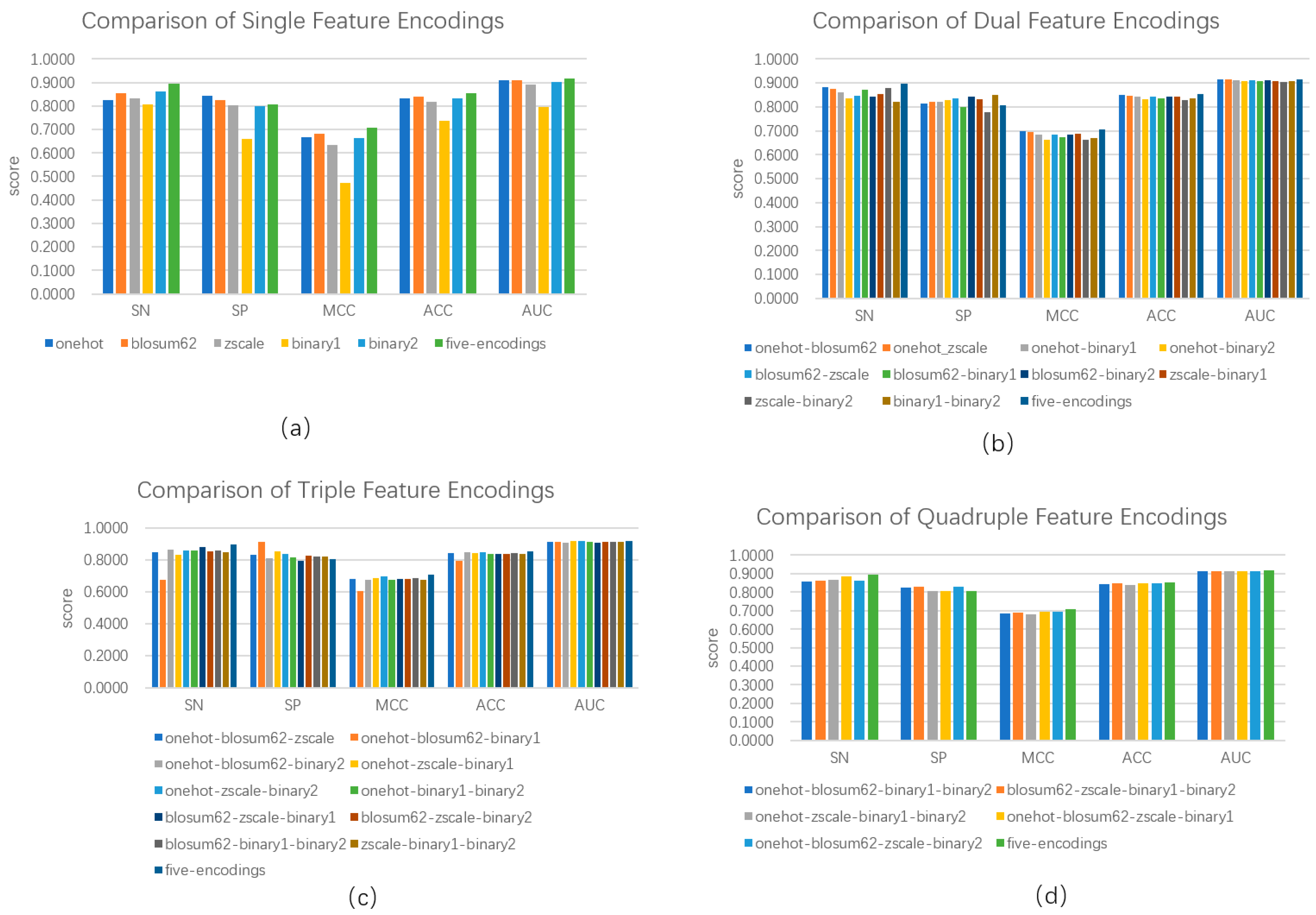
3.2. Feature Selection
3.3. Comparison of Different Structures
3.4. Parameter Optimization
3.5. Comparison with Existing Algorithms
3.6. Comparison with Existing Methods
3.7. Test on Different Positive-to-Negative Sample Ratios
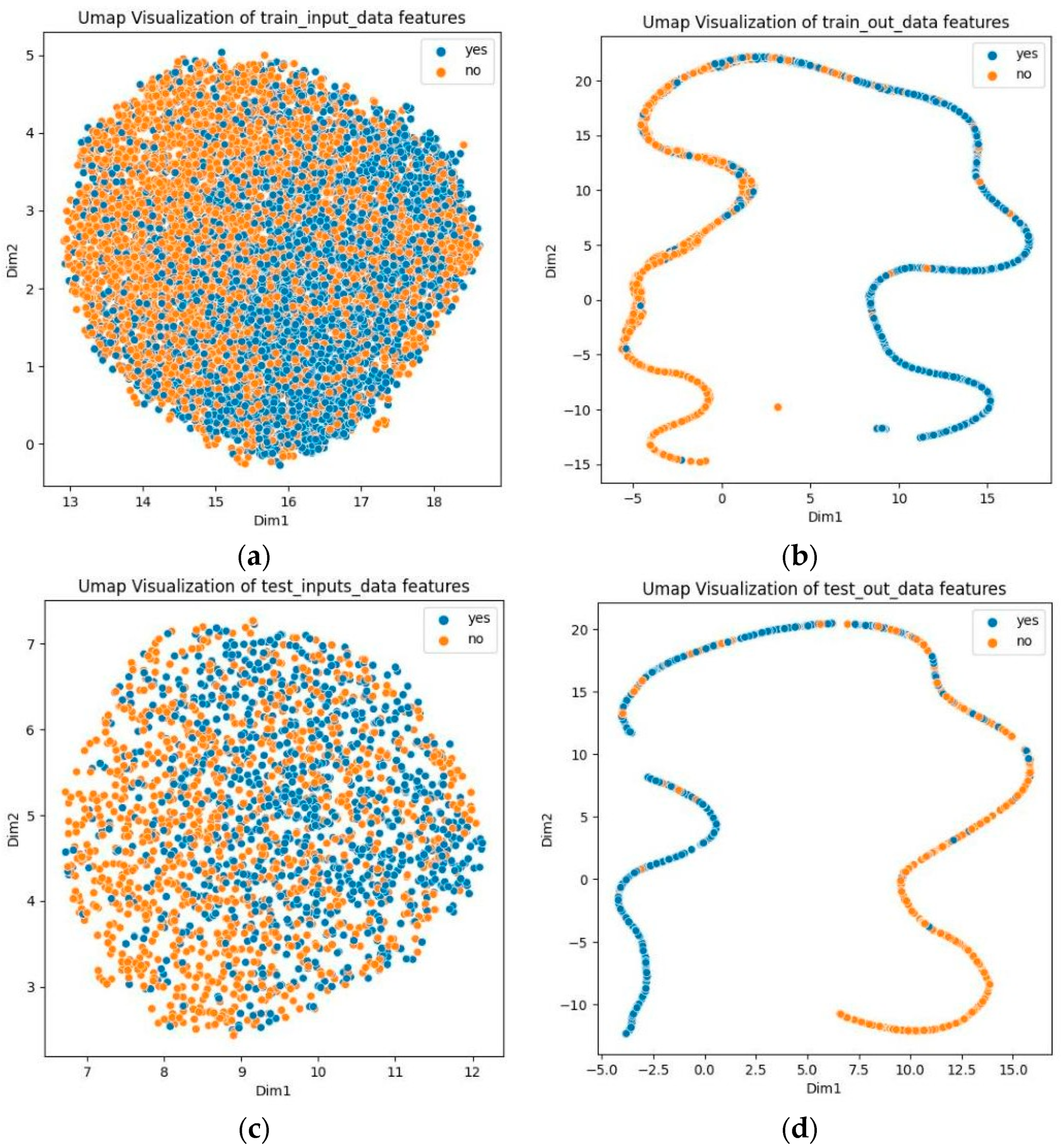
3.8. Visualization Analysis
3.9. GBMPhos Web Server
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cieśla, J.; Frączyk, T.; Rode, W. Phosphorylation of basic amino acid residues in proteins: Important but easily missed. Acta Biochim. Pol. 2011, 58, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Wu, J.; Zou, Q.; Liu, Z.; Xu, L. rBPDL: Predicting RNA-binding proteins using deep learning. IEEE J. Biomed. Health Inform. 2021, 25, 3668–3676. [Google Scholar] [CrossRef]
- Hardman, G.; Perkins, S.; Brownridge, P.J.; Clarke, C.J.; Byrne, D.P.; Campbell, A.E.; Kalyuzhnyy, A.; Myall, A.; Eyers, P.A.; Jones, A.R.; et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 2019, 38, e100847. [Google Scholar] [CrossRef]
- Zhang, W.J.; Zhou, Y.; Zhang, Y.; Su, Y.H.; Xu, T. Protein phosphorylation: A molecular switch in plant signaling. Cell Rep. 2023, 42, 112729. [Google Scholar] [CrossRef]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-κB and IκB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.R.; Veenstra, T.D. Characterization of phosphorylated proteins using mass spectrometry. Curr. Protein Pept. Sci. 2021, 22, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fang, J.; Wang, S.; Zhang, L.; Chen, Y.; Pian, C. Adapt-Kcr: A novel deep learning framework for accurate prediction of lysine crotonylation sites based on learning embedding features and attention architecture. Brief. Bioinform. 2022, 23, bbac037. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Marquez-Lago, T.T.; Leier, A.; Akutsu, T.; Purcell, A.W.; Ian Smith, A.; Lithgow, T.; Daly, R.J.; Song, J.; et al. Quokka: A comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics 2018, 34, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteom. 2008, 7, 1598–1608. [Google Scholar] [CrossRef]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. GPS 5.0: An update on theprediction of kinase-specific phosphorylation sites in proteins. Genom. Proteom. Bioinform. 2020, 18, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.H.; Lee, T.Y.; Liang, H.K.; Huang, C.M.; Wang, T.Y.; Yang, Y.H.; Chu, C.H.; Huang, H.D.; Ko, M.T.; Hwang, J.K. KinasePhos 2.0: A web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 2007, 35, W588–W594. [Google Scholar] [CrossRef]
- Wei, L.; Xing, P.; Tang, J.; Zou, Q. PhosPred-RF: A novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE Trans. Nanobiosci. 2017, 16, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zeng, S.; Xu, C.; Qiu, W.; Liang, Y.; Joshi, T.; Xu, D. MusiteDeep: A deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017, 33, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, D.; Yuchi, J.; He, F.; Jiang, Y.; Cai, S.; Li, J.; Xu, D. MusiteDeep: A deep-learning basedwebserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020, 48, W140–W146. [Google Scholar] [CrossRef]
- Wang, D.; Liang, Y.; Xu, D. Capsule network for protein post-translational modification site prediction. Bioinformatics 2019, 35, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.M.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, Y.; Xu, X.; Cheng, K.K.; Long, Y.; Xu, J.; Li, S.; Dong, J. DeepPSP: A global–local information-based deep neural network for the prediction of protein phosphorylation sites. J. Proteome Res. 2020, 20, 346–356. [Google Scholar] [CrossRef]
- Park, J.H.; Lim, C.Y.; Kwon, H.Y. An experimental study of animating-based facial image manipulation in online class environments. Sci. Rep. 2023, 13, 4667. [Google Scholar] [CrossRef]
- WHO. COVID-19 Weekly Epidemiological Update. [Online]. Available online: https://www.thehinducentre.com/resources/68011999-165.covid-19_epi_update_165.pdf (accessed on 20 May 2024).
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Marrero, M.C.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 2020, 182, 685–712. [Google Scholar] [CrossRef] [PubMed]
- Hekman, R.M.; Hume, A.J.; Goel, R.K.; Abo, K.M.; Huang, J.; Blum, B.C.; Werder, R.B.; Suder, E.L.; Paul, I.; Phanse, S.; et al. Actionable cytopathogenic host responses of human alveolar type 2 cells to SARS-CoV-2. Mol. Cell 2020, 80, 1104–1122. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Dao, F.Y.; Zulfiqar, H.; Lin, H. DeepIPs: Comprehensive assessment and computational identification of phosphorylation sites of SARS-CoV-2 infection using a deep learning-based approach. Brief. Bioinform. 2021, 22, bbab244. [Google Scholar] [CrossRef]
- Zhang, G.; Tang, Q.; Feng, P.; Chen, W. IPs-GRUAtt: An attention-based bidirectional gated recurrent unit network for predicting phosphorylation sites of SARS-CoV-2 infection. Mol. Ther. Nucleic Acids 2023, 32, 28–35. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Zhao, W.; Wu, J.; Luo, S.; Jiang, X.; He, T.; Hu, X. Subtask-aware Representation Learning for Predicting Antibiotic Resistance Gene Properties via Gating-Controlled Mechanism. IEEE J. Biomed. Health Inform. 2024, 28, 4348–4360. [Google Scholar] [CrossRef]
- Xu, J.; Yuan, S.; Shang, J.; Wang, J.; Yan, K.; Yang, Y. Spatiotemporal Network based on GCN and BiGRU for seizure detection. IEEE J. Biomed. Health Inform. 2024, 28, 2037–2046. [Google Scholar] [CrossRef]
- Zhuang, J.; Liu, D.; Lin, M.; Qiu, W.; Liu, J.; Chen, S. PseUdeep: RNA pseudouridine site identification with deep learning algorithm. Front. Genet. 2021, 12, 773882. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, T.; Jiang, Y.; Li, Y.; Li, K.; Quan, L.; Lyu, Q. DeepNup: Prediction of Nucleosome Positioning from DNA Sequences Using Deep Neural Network. Genes 2022, 13, 1983. [Google Scholar] [CrossRef]
- Niu, M.; Lin, Y.; Zou, Q. sgRNACNN: Identifying sgRNA on-target activity in four crops using ensembles of convolutional neural networks. Plant Mol. Biol. 2021, 105, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, C.; Li, F.; Xiang, D.; Chen, Y.Z.; Akutsu, T.; Daly, R.J.; Webb, G.I.; Zhao, Q.; et al. iLearnPlus: A comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021, 49, e60. [Google Scholar] [CrossRef]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, M.; Wold, S. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, X.; Zhang, H. BioSeq-Analysis2.0: An updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 2019, 47, e127. [Google Scholar] [CrossRef]
- White, G.; Seffens, W. Using a neural network to backtranslate amino acid sequences. Electron. J. Biotechnol. 1998, 1, 17–18. [Google Scholar] [CrossRef]
- Lin, K.; May, A.C.; Taylor, W.R. Amino acid encoding schemes from protein structure alignments: Multi-dimensional vectors to describe residue types. J. Theor. Biol. 2002, 216, 361–365. [Google Scholar] [CrossRef]
- Traore, B.B.; Kamsu-Foguem, B.; Tangara, F. Deep convolution neural network for image recognition. Ecol. Inform. 2018, 48, 257–268. [Google Scholar] [CrossRef]
- Yao, G.; Lei, T.; Zhong, J. A review of convolutional-neural-network-based action recognition. Pattern Recognit. Lett. 2019, 118, 14–22. [Google Scholar] [CrossRef]
- Tang, X.; Zheng, P.; Li, X.; Wu, H.; Wei, D.Q.; Liu, Y.; Huang, G. Deep6mAPred: A CNN and Bi-LSTM-based deep learning method for predicting DNA N6-methyladenosine sites across plant species. Methods 2022, 204, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Tayara, H.; Chong, K.T. iPseU-CNN: Identifying RNA pseudouridine sites using convolutional neural networks. Mol. Ther. Nucleic Acids 2019, 16, 463–470. [Google Scholar] [CrossRef]
- Dou, L.; Zhang, Z.; Xu, L.; Zou, Q. iKcr_CNN: A novel computational tool for imbalance classification of human nonhistone crotonylation sites based on convolutional neural networks with focal loss. Comput. Struct. Biotechnol. J. 2022, 20, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Van Merriënboer, B.; Bahdanau, D.; Bengio, Y. On the properties of neural machine translation: Encoder-decoder approaches. arXiv 2014, arXiv:1409.1259. [Google Scholar]
- Huang, G.; Luo, W.; Zhang, G.; Zheng, P.; Yao, Y.; Lyu, J.; Liu, Y.; Wei, D.Q. Enhancer-LSTMAtt: A Bi-LSTM and attention-based deep learning method for enhancer recognition. Biomolecules 2022, 12, 995. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zhang, G.; Liu, Y.; Huang, G. MultiScale-CNN-4mCPred: A multi-scale CNN and adaptive embedding-based method for mouse genome DNA N4-methylcytosine prediction. BMC Bioinform. 2023, 24, 21. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Mikolov, T. Efficient estimation of word representations in vector space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Pennington, J.; Socher, R.; Manning, C.D. Glove: Global vectors for word representation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; pp. 1532–1543. [Google Scholar]
- Bojanowski, P.; Grave, E.; Joulin, A.; Mikolov, T. Enriching word vectors with subword information. Trans. Assoc. Comput. Linguist. 2017, 5, 135–146. [Google Scholar] [CrossRef]
- Joulin, A.; Grave, E.; Bojanowski, P.; Douze, M.; Jégou, H.; Mikolov, T. Fasttext.zip: Compressing text classification models. arXiv 2016, arXiv:1612.03651. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Z1 | Z2 | Z3 | Z4 | Z5 |
|---|---|---|---|---|---|
| A | 0.24 | −2.23 | 0.60 | −0.14 | 1.30 |
| C | 0.84 | −1.67 | 3.71 | 0.18 | −2.65 |
| D | 3.98 | 0.93 | 1.93 | −2.46 | 0.75 |
| E | 3.11 | 0.26 | −0.11 | −3.04 | −0.25 |
| F | −4.22 | 1.94 | 1.06 | 0.54 | −0.62 |
| G | 2.05 | 4.06 | 0.36 | −0.82 | −0.38 |
| H | 2.47 | 1.95 | 0.26 | 3.90 | 0.09 |
| I | −3.89 | −1.73 | −1.71 | −0.84 | 0.26 |
| K | 2.29 | 0.89 | −2.49 | 1.49 | 0.31 |
| L | −4.28 | −1.30 | −1.49 | −0.72 | 0.84 |
| M | −2.85 | −0.22 | 0.47 | 1.94 | −0.98 |
| N | 3.05 | 1.60 | 1.04 | −1.15 | 1.61 |
| P | −1.66 | 0.27 | 1.84 | 0.70 | 2.00 |
| Q | 1.75 | 0.50 | −1.44 | −1.34 | 0.66 |
| R | 3.52 | 2.50 | −3.50 | 1.99 | −0.17 |
| S | 2.39 | −1.07 | 1.15 | −1.39 | 0.67 |
| T | 0.75 | −2.18 | −1.12 | −1.46 | −0.40 |
| V | −2.59 | −2.64 | −1.54 | −0.85 | −0.02 |
| W | −4.36 | 3.94 | 0.59 | 3.44 | −1.59 |
| Y | −2.54 | 2.44 | 0.43 | 0.04 | −1.47 |
| Window Size | SN | SP | ACC | MCC | AUC |
|---|---|---|---|---|---|
| 17 | 0.8253 | 0.7997 | 0.8131 | 0.6253 | 0.8785 |
| 19 | 0.8195 | 0.7922 | 0.8065 | 0.612 | 0.8814 |
| 21 | 0.8655 | 0.7657 | 0.8179 | 0.6359 | 0.8875 |
| 23 | 0.8230 | 0.8401 | 0.8311 | 0.6624 | 0.8948 |
| 25 | 0.8460 | 0.8111 | 0.8293 | 0.6578 | 0.9028 |
| 27 | 0.8494 | 0.8136 | 0.8323 | 0.6638 | 0.9005 |
| 29 | 0.8667 | 0.7746 | 0.8227 | 0.6453 | 0.8957 |
| 31 | 0.8448 | 0.8186 | 0.8323 | 0.6638 | 0.9038 |
| 33 | 0.8953 | 0.8072 | 0.8528 | 0.7066 | 0.9163 |
| Method | SN | SP | ACC | MCC | AUC |
|---|---|---|---|---|---|
| BiLSTM | 0.8582 | 0.8109 | 0.8347 | 0.6700 | 0.9103 |
| Without Bi-GRU | 0 | 1 | 0.4832 | nan | 0.5000 |
| Without Conv1 | 0.8558 | 0.8221 | 0.8395 | 0.6786 | 0.9129 |
| Without Conv2 | 0.8640 | 0.8097 | 0.8377 | 0.6753 | 0.9076 |
| Without gating | 0.8349 | 0.8420 | 0.8383 | 0.6766 | 0.9113 |
| Bi-GRU | 0.8953 | 0.8072 | 0.8528 | 0.7066 | 0.9163 |
| Method | SN | SP | ACC | MCC | AUC |
|---|---|---|---|---|---|
| Conv1 (1) | 0.8953 | 0.8072 | 0.8528 | 0.7066 | 0.9163 |
| Conv1 (3) | 0.8535 | 0.8231 | 0.8383 | 0.6762 | 0.9071 |
| Conv1 (5) | 0.8756 | 0.7823 | 0.8305 | 0.6620 | 0.9033 |
| Conv1 (7) | 0.8558 | 0.8010 | 0.8293 | 0.6584 | 0.9030 |
| Conv2 (3, 5, 7) | 0.8674 | 0.8122 | 0.8407 | 0.6813 | 0.9108 |
| Conv2 (1, 3, 5) | 0.8663 | 0.8134 | 0.8407 | 0.6813 | 0.9095 |
| Conv2 (5, 7, 9) | 0.8872 | 0.8122 | 0.8510 | 0.7024 | 0.9167 |
| Conv2 (7, 9, 11) | 0.8140 | 0.8619 | 0.8371 | 0.6757 | 0.9103 |
| Conv2 (1, 1, 1) | 0.8415 | 0.8267 | 0.8341 | 0.6683 | 0.9051 |
| Conv2 (5, 5, 5) | 0.8391 | 0.8255 | 0.8323 | 0.6646 | 0.9023 |
| Conv2 (7, 7, 7) | 0.8117 | 0.8485 | 0.8299 | 0.6605 | 0.8941 |
| Conv2 (3, 3, 3) | 0.8953 | 0.8072 | 0.8528 | 0.7066 | 0.9163 |
| Method | SN | SP | ACC | MCC | AUC |
|---|---|---|---|---|---|
| LR | 0.8004 | 0.7770 | 0.7884 | 0.5772 | 0.8687 |
| DT | 0.7120 | 0.6736 | 0.6923 | 0.3857 | 0.6928 |
| SVM | 0.8089 | 0.7987 | 0.8037 | 0.6075 | 0.8827 |
| RF | 0.7243 | 0.8531 | 0.7903 | 0.5832 | 0.8715 |
| GBDT | 0.7861 | 0.8286 | 0.8079 | 0.6156 | 0.8916 |
| XGB | 0.8108 | 0.8178 | 0.8144 | 0.6286 | 0.8928 |
| LGBM | 0.8146 | 0.8259 | 0.8204 | 0.6406 | 0.9035 |
| GBMPhos | 0.8513 | 0.8500 | 0.8506 | 0.7010 | 0.9209 |
| Method | SN | SP | ACC | MCC | AUC |
|---|---|---|---|---|---|
| DeepIPs | 0.8007 | 0.8109 | 0.8059 | 0.6117 | 0.8926 |
| Adapt-Kcr | 0.8090 | 0.8572 | 0.8332 | 0.6670 | 0.9120 |
| IPs-GRUAtt | 0.8378 | 0.8545 | 0.8462 | 0.6924 | 0.9187 |
| GBMPhos | 0.8513 | 0.8500 | 0.8506 | 0.7010 | 0.9209 |
| Method | SN | SP | ACC | MCC | AUC |
|---|---|---|---|---|---|
| DeepIPs | 0.9048 | 0.8095 | 0.8333 | 0.7175 | 0.9252 |
| IPs-GRUAtt | 0.9524 | 0.9048 | 0.9286 | 0.8581 | 0.9206 |
| DeepPSP | 0.9524 | 0.5714 | 0.7619 | 0.5665 | 0.8209 |
| GBMPhos | 0.9333 | 0.8800 | 0.9000 | 0.7965 | 0.9000 |
| Phosphorylated S/T Sites | Method | Predicted Sites |
|---|---|---|
| 5, 161, 163, 198, 201, 295, 334, 728, 735, 806, 975, 1035 | MusiteDeep | 96, 163, 198, 201, 555, 678, 749, 752, 975, 1035 |
| DeepIPs | 102, 163, 198, 201, 334, 387, 740, 742, 1016 | |
| IPs-GRUAtt | 102, 198, 201, 387, 680 | |
| GBMPhos | 102, 163, 198, 201, 334, 387, 680 |
| Ratio | SP | ACC | AUC | AUPRC |
|---|---|---|---|---|
| 1:1 | 0.8500 | 0.8506 | 0.9209 | 0.9245 |
| 1:2 | 0.8208 | 0.8303 | 0.9025 | 0.8119 |
| 1:3 | 0.8152 | 0.8235 | 0.9018 | 0.7507 |
| 1:4 | 0.8111 | 0.8186 | 0.9007 | 0.7051 |
| 1:5 | 0.8086 | 0.8153 | 0.8988 | 0.6574 |
| 1:6 | 0.8051 | 0.8113 | 0.8974 | 0.6232 |
| 1:7 | 0.8016 | 0.8074 | 0.8964 | 0.5887 |
| 1:8 | 0.7977 | 0.8033 | 0.8956 | 0.5608 |
| 1:9 | 0.794 | 0.7994 | 0.8946 | 0.5386 |
| 1:10 | 0.7898 | 0.7950 | 0.8934 | 0.5129 |
| Mean and Standard Deviation | 0.8049 ± 0.0101 | 0.8116 ± 0.0115 | 0.8979 ± 0.0032 | 0.6674 ± 0.1314 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, G.; Xiao, R.; Chen, W.; Dai, Q. GBMPhos: A Gating Mechanism and Bi-GRU-Based Method for Identifying Phosphorylation Sites of SARS-CoV-2 Infection. Biology 2024, 13, 798. https://doi.org/10.3390/biology13100798
Huang G, Xiao R, Chen W, Dai Q. GBMPhos: A Gating Mechanism and Bi-GRU-Based Method for Identifying Phosphorylation Sites of SARS-CoV-2 Infection. Biology. 2024; 13(10):798. https://doi.org/10.3390/biology13100798
Chicago/Turabian StyleHuang, Guohua, Runjuan Xiao, Weihong Chen, and Qi Dai. 2024. "GBMPhos: A Gating Mechanism and Bi-GRU-Based Method for Identifying Phosphorylation Sites of SARS-CoV-2 Infection" Biology 13, no. 10: 798. https://doi.org/10.3390/biology13100798