
Ablation of TrkB from Enkephalinergic Precursor-Derived Cerebellar Granule Cells Generates Ataxia

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Tissue Processing
2.3. Immunofluorescence
2.4. Estimation of a Proportion of GCs and PCs Undergoing Recombination
2.5. PC Count Analysis
2.6. Nissl Staining and Cavalieri Analysis
2.7. Immunoblotting
2.8. Behavioural Analysis
2.8.1. Hindlimb Clasping
2.8.2. Kyphosis Test
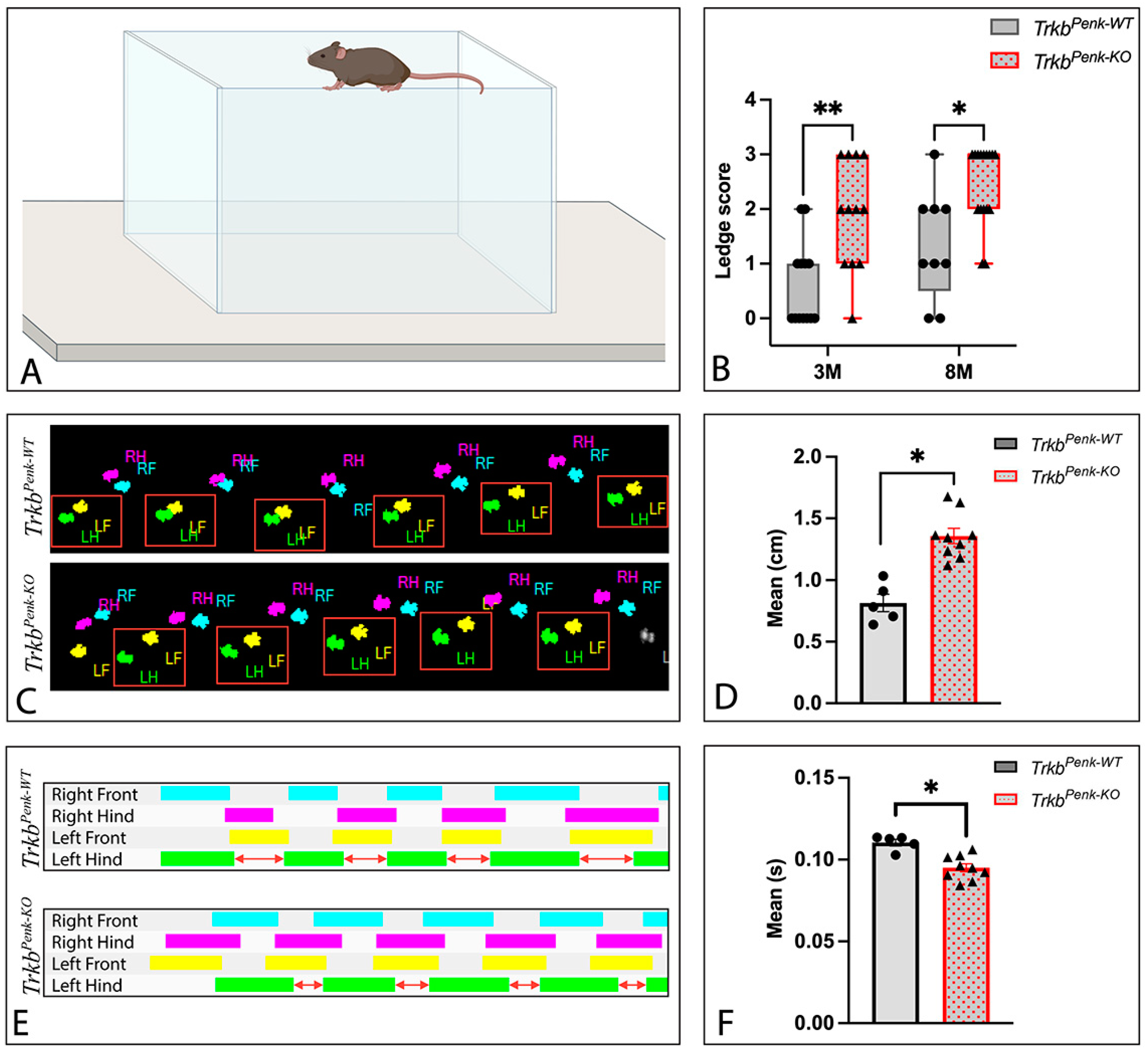
2.8.3. Ledge Test
2.8.4. Catwalk Test
2.9. Statistical Analysis
3. Results
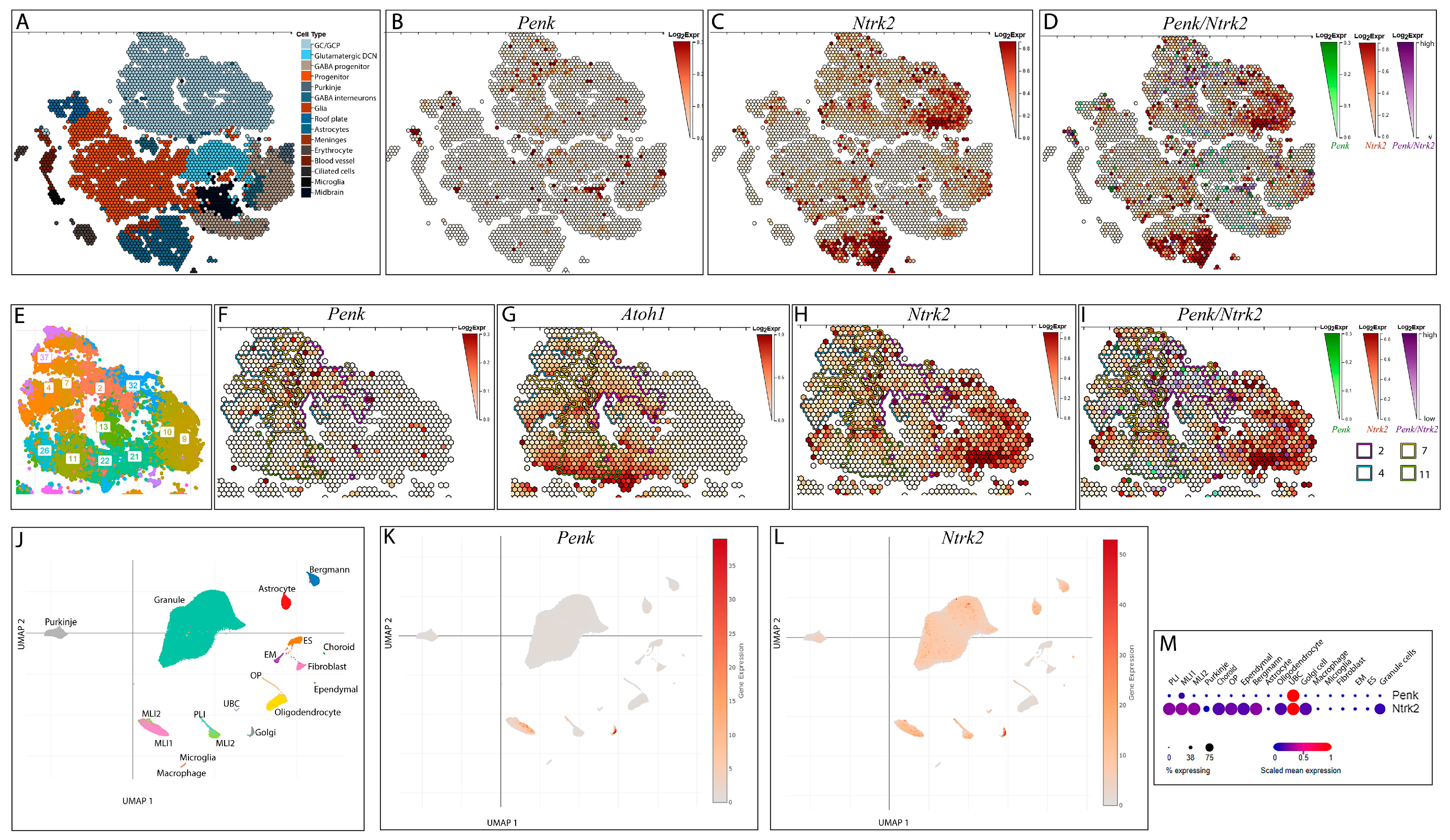
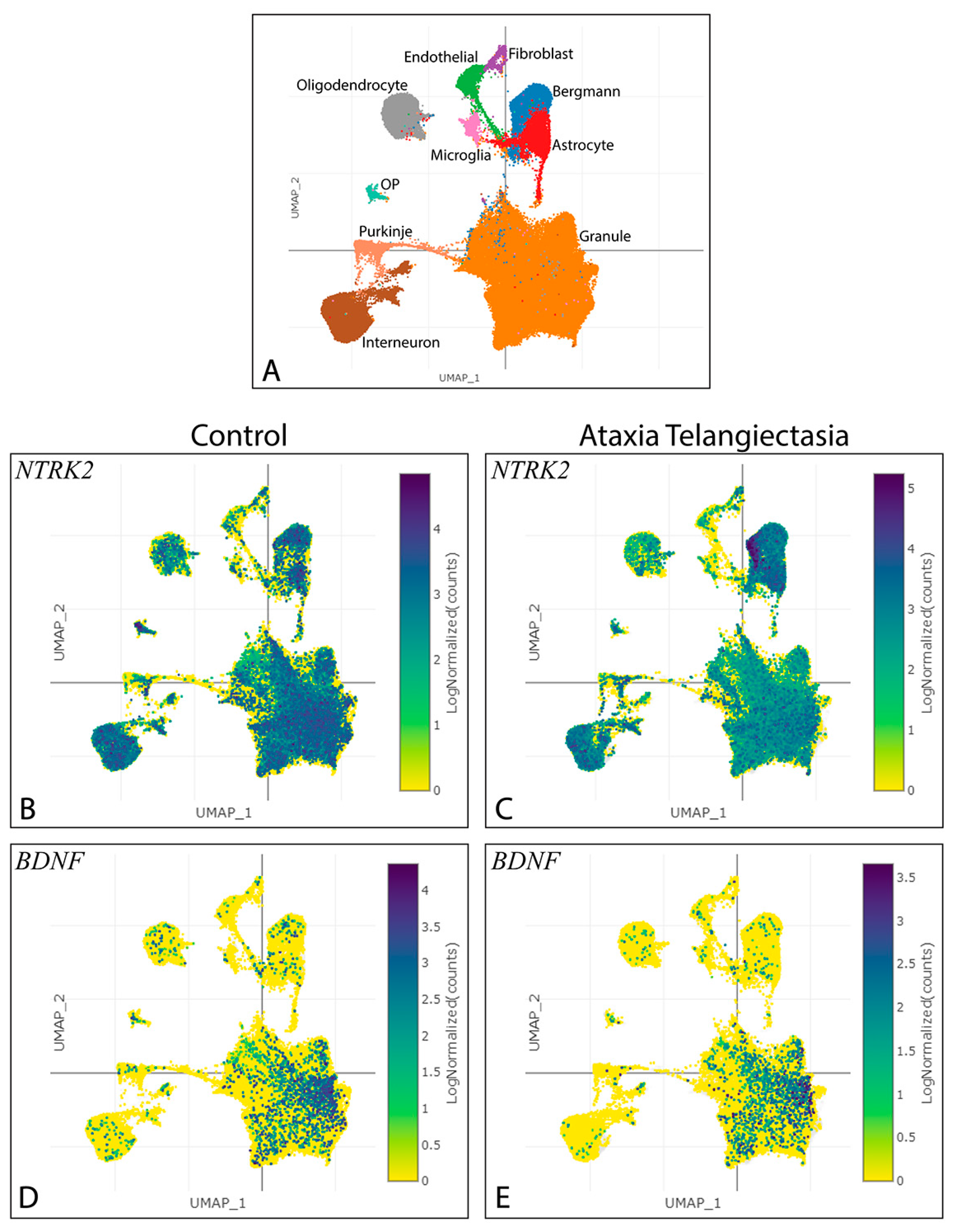
3.1. Exploring the Impact of Ntrk2 Deletion in a Specific Subset of Cerebellar Granule Cells
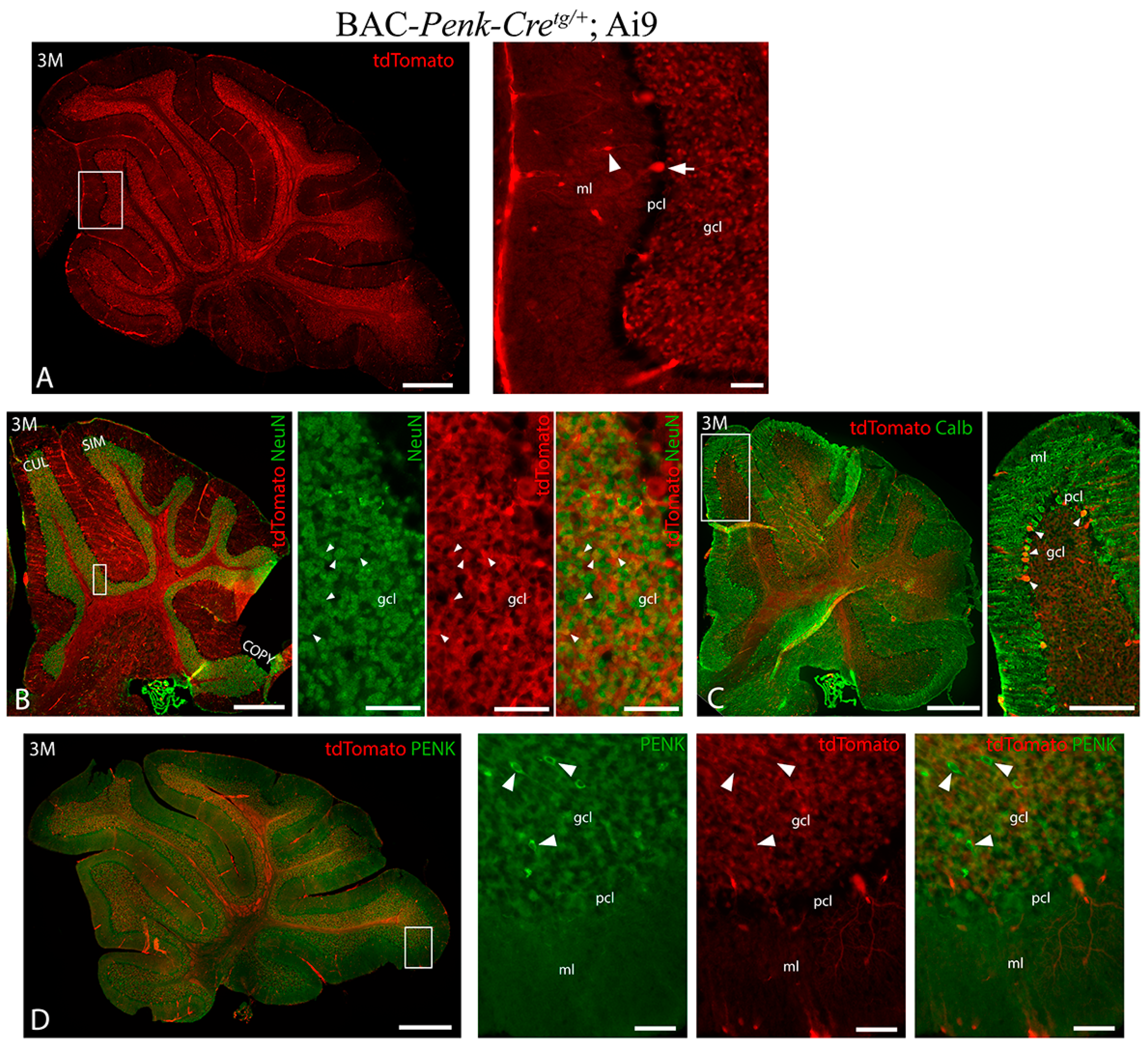
3.2. Pattern of BAC-Penk-Cre Mediated Recombination in the Cerebellum
3.3. Unaltered Cerebellar Morphology of TrkbPenk-KO Mice
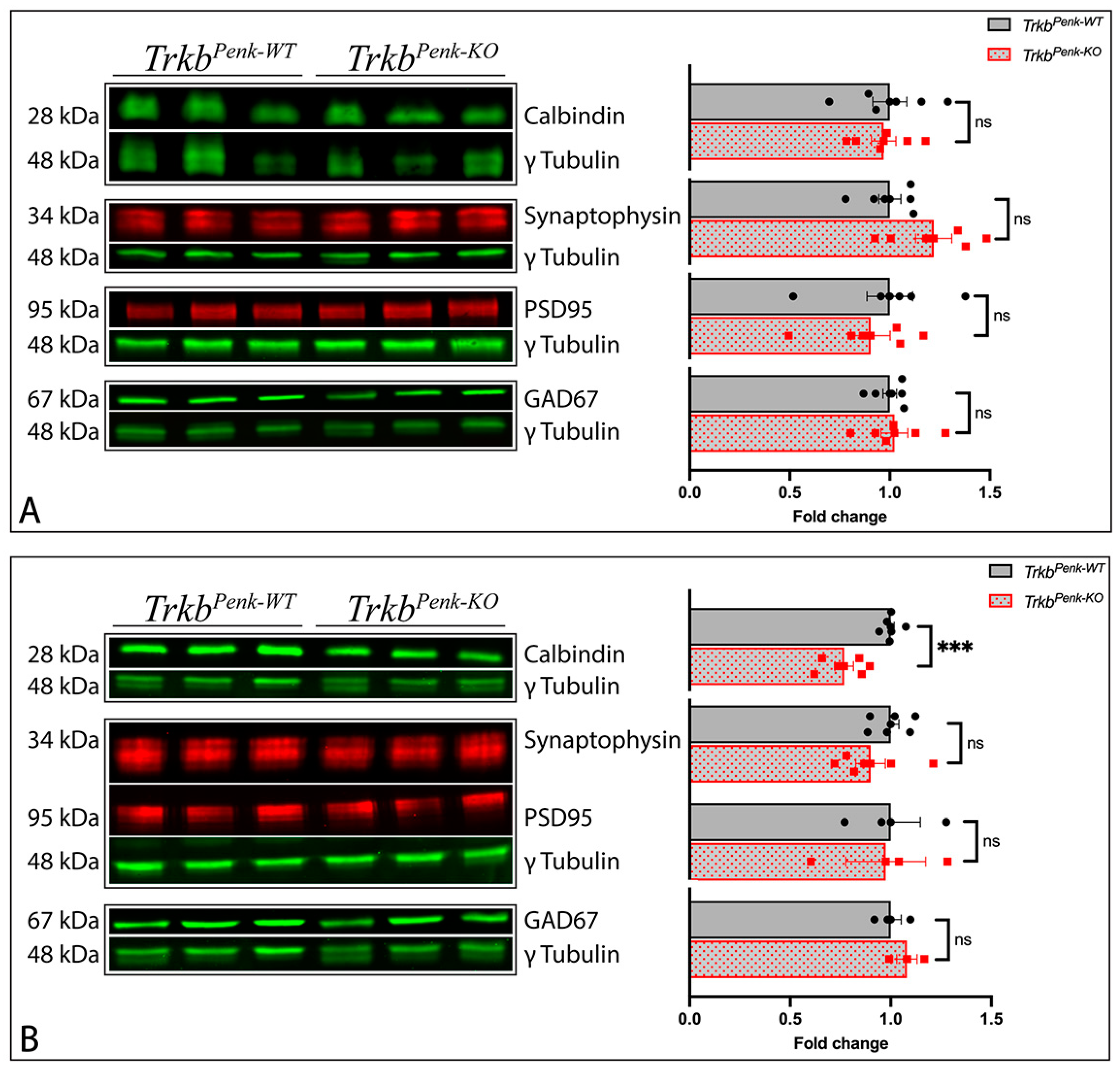
3.4. Molecular Consequences of Ntrk2 Deletion in TrkbPenk-KO Mice
3.5. Behavioural Consequences of Ntrk2 Deletion in Enk-Derived GCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bastian, A.J. Mechanisms of Ataxia. Phys. Ther. 1997, 77, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Seidel, K.; Siswanto, S.; Brunt, E.R.P.; den Dunnen, W.; Korf, H.-W.; Rüb, U. Brain Pathology of Spinocerebellar Ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Verbeek, D.S. Why Do so Many Genetic Insults Lead to Purkinje Cell Degeneration and Spinocerebellar Ataxia? Neurosci. Lett. 2019, 688, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, E.; Balbo, I.; Miniaci, M.C.; Tempia, F. Purkinje Cell Signaling Deficits in Animal Models of Ataxia. Front. Synaptic Neurosci. 2018, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Ishikawa, K.; Sato, N.; Obayashi, M.; Niimi, Y.; Ishiguro, T.; Yamada, M.; Toyoshima, Y.; Takahashi, H.; Kato, T.; et al. Reduced Brain-Derived Neurotrophic Factor (BDNF) mRNA Expression and Presence of BDNF-Immunoreactive Granules in the Spinocerebellar Ataxia Type 6 (SCA6) Cerebellum. Neuropathology 2012, 32, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Misiorek, J.O.; Schreiber, A.M.; Urbanek-Trzeciak, M.O.; Jazurek-Ciesiołka, M.; Hauser, L.A.; Lynch, D.R.; Napierala, J.S.; Napierala, M. A Comprehensive Transcriptome Analysis Identifies FXN and BDNF as Novel Targets of miRNAs in Friedreich’s Ataxia Patients. Mol. Neurobiol. 2020, 57, 2639–2653. [Google Scholar] [CrossRef] [PubMed]
- Rosa, J.-G.; Hamel, K.; Soles, A.; Sheeler, C.; Borgenheimer, E.; Gilliat, S.; Sbrocco, K.; Ghanoum, F.; Handler, H.P.; Forster, C.; et al. BDNF Is Altered in a Brain-Region Specific Manner and Rescues Deficits in Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2023, 178, 106023. [Google Scholar] [CrossRef] [PubMed]
- Mellesmoen, A.; Sheeler, C.; Ferro, A.; Rainwater, O.; Cvetanovic, M. Brain Derived Neurotrophic Factor (BDNF) Delays Onset of Pathogenesis in Transgenic Mouse Model of Spinocerebellar Ataxia Type 1 (SCA1). Front. Cell. Neurosci. 2019, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sheeler, C.; Rosa, J.-G.; Borgenheimer, E.; Mellesmoen, A.; Rainwater, O.; Cvetanovic, M. Post-Symptomatic Delivery of Brain-Derived Neurotrophic Factor (BDNF) Ameliorates Spinocerebellar Ataxia Type 1 (SCA1) Pathogenesis. Cerebellum 2021, 20, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.A.; Jayabal, S.; Sheng, J.; Fields, E.; Leung, T.C.S.; Quilez, S.; McNicholas, E.; Lau, L.; Huang, S.; Watt, A.J. Activation of TrkB-Akt Signaling Rescues Deficits in a Mouse Model of SCA6. Sci. Adv. 2022, 8, eabh3260. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Demirbas, D.; Kim, J.; Jeffries, A.M.; Tolles, A.; Park, J.; Chittenden, T.W.; Buckley, P.G.; Yu, T.W.; Lodato, M.A.; et al. ATM-Deficiency-Induced Microglial Activation Promotes Neurodegeneration in Ataxia-Telangiectasia. Cell Rep. 2024, 43. [Google Scholar] [CrossRef] [PubMed]
- Kozareva, V.; Martin, C.; Osorno, T.; Rudolph, S.; Guo, C.; Vanderburg, C.; Nadaf, N.; Regev, A.; Regehr, W.G.; Macosko, E. A Transcriptomic Atlas of Mouse Cerebellar Cortex Comprehensively Defines Cell Types. Nature 2021, 598, 214–219. [Google Scholar] [CrossRef]
- Huang, C.; Gammon, S.J.; Dieterle, M.; Huang, R.H.; Likins, L.; Ricklefs, R.E. Dramatic Increases in Number of Cerebellar Granule-Cell-Purkinje-Cell Synapses across Several Mammals. Mamm. Biol. 2014, 79, 163–169. [Google Scholar] [CrossRef]
- Bradley, P.; Berry, M. The Effects of Reduced Climbing and Parallel Fibre Input on Purkinje Cell Dendritic Growth. Brain Res. 1976, 109, 133–151. [Google Scholar] [CrossRef]
- Hashimoto, K.; Yoshida, T.; Sakimura, K.; Mishina, M.; Watanabe, M.; Kano, M. Influence of Parallel Fiber–Purkinje Cell Synapse Formation on Postnatal Development of Climbing Fiber–Purkinje Cell Synapses in the Cerebellum. Neuroscience 2009, 162, 601–611. [Google Scholar] [CrossRef]
- Galliano, E.; Gao, Z.; Schonewille, M.; Todorov, B.; Simons, E.; Pop, A.S.; D’Angelo, E.; van den Maagdenberg, A.M.J.M.; Hoebeek, F.E.; De Zeeuw, C.I. Silencing the Majority of Cerebellar Granule Cells Uncovers Their Essential Role in Motor Learning and Consolidation. Cell Rep. 2013, 3, 1239–1251. [Google Scholar] [CrossRef]
- Besusso, D.; Geibel, M.; Kramer, D.; Schneider, T.; Pendolino, V.; Picconi, B.; Calabresi, P.; Bannerman, D.M.; Minichiello, L. BDNF–TrkB Signaling in Striatopallidal Neurons Controls Inhibition of Locomotor Behavior. Nat. Commun. 2013, 4, 2031. [Google Scholar] [CrossRef]
- Zagon, I.S.; McLaughlin, P.J. Ultrastructural Localization of Enkephalin-like Immunoreactivity in Developing Rat Cerebellum. Neuroscience 1990, 34, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.G.; Kindy, M.S.; Spruce, B.A.; Hauser, K.F. Ontogeny of Proenkephalin mRNA and Enkephalin Peptide Expression in the Cerebellar Cortex of the Rat: Spatial and Temporal Patterns of Expression Follow Maturational Gradients in the External Granular Layer and in Purkinje Cells. Dev. Brain Res. 1993, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Rhodes, R.E.; McLaughlin, P.J. Distribution of Enkephalin Immunoreactivity in Germinative Cells of Developing Rat Cerebellum. Science 1985, 227, 1049–1051. [Google Scholar] [CrossRef]
- Carter, R.A.; Bihannic, L.; Rosencrance, C.; Hadley, J.L.; Tong, Y.; Phoenix, T.N.; Natarajan, S.; Easton, J.; Northcott, P.A.; Gawad, C. A Single-Cell Transcriptional Atlas of the Developing Murine Cerebellum. Curr. Biol. 2018, 28, 2910–2920.e2. [Google Scholar] [CrossRef]
- Minichiello, L.; Korte, M.; Wolfer, D.; Kühn, R.; Unsicker, K.; Cestari, V.; Rossi-Arnaud, C.; Lipp, H.-P.; Bonhoeffer, T.; Klein, R. Essential Role for TrkB Receptors in Hippocampus-Mediated Learning. Neuron 1999, 24, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A Robust and High-Throughput Cre Reporting and Characterization System for the Whole Mouse Brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Minichiello, L.; Besusso, D.; Geibel, M. Immunohistochemical Detection of Preproenkephalin in the Mouse Brain. Available online: https://protocolexchange.researchsquare.com/article/nprot-2738/v1 (accessed on 11 August 2023).
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Sahin, B.; Emirzeoglu, M.; Uzun, A.; Incesu, L.; Bek, Y.; Bilgic, S.; Kaplan, S. Unbiased Estimation of the Liver Volume by the Cavalieri Principle Using Magnetic Resonance Images. Eur. J. Radiol. 2003, 47, 164–170. [Google Scholar] [CrossRef]
- Chou, A.-H.; Yeh, T.-H.; Ouyang, P.; Chen, Y.-L.; Chen, S.-Y.; Wang, H.-L. Polyglutamine-Expanded Ataxin-3 Causes Cerebellar Dysfunction of SCA3 Transgenic Mice by Inducing Transcriptional Dysregulation. Neurobiol. Dis. 2008, 31, 89–101. [Google Scholar] [CrossRef]
- Guyenet, S.J.; Furrer, S.A.; Damian, V.M.; Baughan, T.D.; La Spada, A.R.; Garden, G.A. A Simple Composite Phenotype Scoring System for Evaluating Mouse Models of Cerebellar Ataxia. JoVE J. Vis. Exp. 2010, 39, e1787. [Google Scholar] [CrossRef]
- Caballero-Garrido, E.; Pena-Philippides, J.C.; Galochkina, Z.; Erhardt, E.; Roitbak, T. Characterization of Long-Term Gait Deficits in Mouse dMCAO, Using the CatWalk System. Behav. Brain Res. 2017, 331, 282–296. [Google Scholar] [CrossRef]
- Timotius, I.K.; Roelofs, R.F.; Richmond-Hacham, B.; Noldus, L.P.J.J.; von Hörsten, S.; Bikovski, L. CatWalk XT Gait Parameters: A Review of Reported Parameters in Pre-Clinical Studies of Multiple Central Nervous System and Peripheral Nervous System Disease Models. Front. Behav. Neurosci. 2023, 17, 1147784. [Google Scholar] [CrossRef]
- Khouri-Farah, N.; Guo, Q.; Morgan, K.; Shin, J.; Li, J.Y.H. Integrated Single-Cell Transcriptomic and Epigenetic Study of Cell State Transition and Lineage Commitment in Embryonic Mouse Cerebellum. Sci. Adv. 2022, 8, eabl9156. [Google Scholar] [CrossRef]
- Blum, J.A.; Klemm, S.; Shadrach, J.L.; Guttenplan, K.A.; Nakayama, L.; Kathiria, A.; Hoang, P.T.; Gautier, O.; Kaltschmidt, J.A.; Greenleaf, W.J.; et al. Single-Cell Transcriptomic Analysis of the Adult Mouse Spinal Cord Reveals Molecular Diversity of Autonomic and Skeletal Motor Neurons. Nat. Neurosci. 2021, 24, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Ben-Arie, N.; Bellen, H.J.; Armstrong, D.L.; McCall, A.E.; Gordadze, P.R.; Guo, Q.; Matzuk, M.M.; Zoghbi, H.Y. Math1 Is Essential for Genesis of Cerebellar Granule Neurons. Nature 1997, 390, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Weyer, A.; Schilling, K. Developmental and cell type-specific expression of the neuronal marker NeuN in the murine cerebellum. J. Neurosci. Res. 2003, 73, 400–409. [Google Scholar] [CrossRef] [PubMed]
- François, A.; Low, S.A.; Sypek, E.I.; Christensen, A.J.; Sotoudeh, C.; Beier, K.T.; Ramakrishnan, C.; Ritola, K.D.; Sharif-Naeini, R.; Deisseroth, K.; et al. A Brainstem-Spinal Cord Inhibitory Circuit for Mechanical Pain Modulation by GABA and Enkephalins. Neuron 2017, 93, 822–839.e6. [Google Scholar] [CrossRef]
- Fukushima, T.; Tsuda, M.; Kofuji, T.; Hori, Y. Physiological Properties of Enkephalin-Containing Neurons in the Spinal Dorsal Horn Visualized by Expression of Green Fluorescent Protein in BAC Transgenic Mice. BMC Neurosci. 2011, 12, 36. [Google Scholar] [CrossRef]
- Pezet, S.; Cunningham, J.; Patel, J.; Grist, J.; Gavazzi, I.; Lever, I.J.; Malcangio, M. BDNF Modulates Sensory Neuron Synaptic Activity by a Facilitation of GABA Transmission in the Dorsal Horn. Mol. Cell. Neurosci. 2002, 21, 51–62. [Google Scholar] [CrossRef]
- Alkaslasi, M.R.; Piccus, Z.E.; Hareendran, S.; Silberberg, H.; Chen, L.; Zhang, Y.; Petros, T.J.; Le Pichon, C.E. Single Nucleus RNA-Sequencing Defines Unexpected Diversity of Cholinergic Neuron Types in the Adult Mouse Spinal Cord. Nat. Commun. 2021, 12, 2471. [Google Scholar] [CrossRef]
- Rico, B.; Xu, B.; Reichardt, L.F. TrkB Receptor Signaling Is Required for Establishment of GABAergic Synapses in the Cerebellum. Nat. Neurosci. 2002, 5, 225–233. [Google Scholar] [CrossRef]
- Chen, A.I.; Nguyen, C.N.; Copenhagen, D.R.; Badurek, S.; Minichiello, L.; Ranscht, B.; Reichardt, L.F. TrkB (Tropomyosin-Related Kinase B) Controls the Assembly and Maintenance of GABAergic Synapses in the Cerebellar Cortex. J. Neurosci. 2011, 31, 2769–2780. [Google Scholar] [CrossRef]
- Buckley, E.; Mazzà, C.; McNeill, A. A Systematic Review of the Gait Characteristics Associated with Cerebellar Ataxia. Gait Posture 2018, 60, 154–163. [Google Scholar] [CrossRef]
- Kim, J.C.; Cook, M.N.; Carey, M.R.; Shen, C.; Regehr, W.G.; Dymecki, S.M. Linking Genetically Defined Neurons to Behavior through a Broadly Applicable Silencing Allele. Neuron 2009, 63, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, T.; Kim, J.; Yamamoto, Y.; Tanaka-Yamamoto, K. Inputs from Sequentially Developed Parallel Fibers Are Required for Cerebellar Organization. Cell Rep. 2019, 28, 2939–2954.e5. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Khan, M.M.; Stark, A.P.; Seo, S.; Norton, A.; Yao, Z.; Chen, C.H.; Regehr, W.G. Cerebellar Granule Cell Signaling Is Indispensable for Normal Motor Performance. Cell Rep. 2023, 42, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal Dominant Cerebellar Ataxia (SCA6) Associated with Small Polyglutamine Expansions in the α1A-Voltage-Dependent Calcium Channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Tanaka, O.; Sakagami, H.; Kondo, H. Localization of mRNAs of Voltage-Dependent Ca2+-Channels: Four Subtypes of A1- and β-Subunits in Developing and Mature Rat Brain. Mol. Brain Res. 1995, 30, 1–16. [Google Scholar] [CrossRef]
- Ishikawa, K.; Watanabe, M.; Yoshizawa, K.; Fujita, T.; Iwamoto, H.; Yoshizawa, T.; Harada, K.; Nakamagoe, K.; Komatsuzaki, Y.; Satoh, A.; et al. Clinical, Neuropathological, and Molecular Study in Two Families with Spinocerebellar Ataxia Type 6 (SCA6). J. Neurol. Neurosurg. Psychiatry 1999, 67, 86–89. [Google Scholar] [CrossRef]
- Takahashi, H.; Ikeuchi, T.; Honma, Y.; Hayashi, S.; Tsuji, S. Autosomal Dominant Cerebellar Ataxia (SCA6): Clinical, Genetic and Neuropathological Study in a Family. Acta Neuropathol. 1998, 95, 333–337. [Google Scholar] [CrossRef]
- Watase, K.; Barrett, C.F.; Miyazaki, T.; Ishiguro, T.; Ishikawa, K.; Hu, Y.; Unno, T.; Sun, Y.; Kasai, S.; Watanabe, M.; et al. Spinocerebellar Ataxia Type 6 Knockin Mice Develop a Progressive Neuronal Dysfunction with Age-Dependent Accumulation of Mutant CaV2.1 Channels. Proc. Natl. Acad. Sci. USA 2008, 105, 11987–11992. [Google Scholar] [CrossRef]
- Jayabal, S.; Ljungberg, L.; Erwes, T.; Cormier, A.; Quilez, S.; Jaouhari, S.E.; Watt, A.J. Rapid Onset of Motor Deficits in a Mouse Model of Spinocerebellar Ataxia Type 6 Precedes Late Cerebellar Degeneration. eNeuro 2015, 2, 1–18. [Google Scholar] [CrossRef]
- Jayabal, S.; Chang, H.H.V.; Cullen, K.E.; Watt, A.J. 4-Aminopyridine Reverses Ataxia and Cerebellar Firing Deficiency in a Mouse Model of Spinocerebellar Ataxia Type 6. Sci. Rep. 2016, 6, 29489. [Google Scholar] [CrossRef]
- Jayabal, S.; Ljungberg, L.; Watt, A.J. Transient Cerebellar Alterations during Development Prior to Obvious Motor Phenotype in a Mouse Model of Spinocerebellar Ataxia Type 6. J. Physiol. 2017, 595, 949–966. [Google Scholar] [CrossRef] [PubMed]
- Unno, T.; Wakamori, M.; Koike, M.; Uchiyama, Y.; Ishikawa, K.; Kubota, H.; Yoshida, T.; Sasakawa, H.; Peters, C.; Mizusawa, H.; et al. Development of Purkinje Cell Degeneration in a Knockin Mouse Model Reveals Lysosomal Involvement in the Pathogenesis of SCA6. Proc. Natl. Acad. Sci. USA 2012, 109, 17693–17698. [Google Scholar] [CrossRef] [PubMed]
- Mark, M.D.; Krause, M.; Boele, H.-J.; Kruse, W.; Pollok, S.; Kuner, T.; Dalkara, D.; Koekkoek, S.; Zeeuw, C.I.D.; Herlitze, S. Spinocerebellar Ataxia Type 6 Protein Aggregates Cause Deficits in Motor Learning and Cerebellar Plasticity. J. Neurosci. 2015, 35, 8882–8895. [Google Scholar] [CrossRef]
- Johnson, E.M.; Craig, E.T.; Yeh, H.H. TrkB Is Necessary for Pruning at the Climbing Fibre–Purkinje Cell Synapse in the Developing Murine Cerebellum. J. Physiol. 2007, 582, 629–646. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Reichardt, L.F. Cre-Mediated Recombination in Rhombic Lip Derivatives. Genesis 2002, 33, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.I.; Zang, K.; Masliah, E.; Reichardt, L.F. Glutamatergic Axon-Derived BDNF Controls GABAergic Synaptic Differentiation in the Cerebellum. Sci. Rep. 2016, 6, 20201. [Google Scholar] [CrossRef]
- Zagon, I.S.; Isayama, T.; McLaughlin, P.J. Preproenkephalin mRNA Expression in the Developing and Adult Rat Brain. Mol. Brain Res. 1994, 21, 85–98. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Primers 2015, 1, 1–21. [Google Scholar] [CrossRef]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. RESEARCH ARTICLE: Systematic Assessment of BDNF and Its Receptor Levels in Human Cortices Affected by Huntington’s Disease. Brain Pathol. 2008, 18, 225–238. [Google Scholar] [CrossRef]
- Nguyen, H.; Ostendorf, A.P.; Satz, J.S.; Westra, S.; Ross-Barta, S.E.; Campbell, K.P.; Moore, S.A. Glial Scaffold Required for Cerebellar Granule Cell Migration Is Dependent on Dystroglycan Function as a Receptor for Basement Membrane Proteins. Acta Neuropathol. Commun. 2013, 1, 58. [Google Scholar] [CrossRef]
- Franklin, G.L.; Camargo, C.H.F.; Meira, A.T.; Pavanelli, G.M.; Milano, S.S.; Germiniani, F.B.; Lima, N.S.C.; Raskin, S.; Barsottini, O.G.P.; Pedroso, J.L.; et al. Is Ataxia an Underestimated Symptom of Huntington’s Disease? Front. Neurol. 2020, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.K.; Muratori, L.; Louis, E.D.; Moskowitz, C.B.; Marder, K.S. Spectrum of Gait Impairments in Presymptomatic and Symptomatic Huntington’s Disease. Mov. Disord. 2008, 23, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Latimer, C.S.; Flanagan, M.E.; Cimino, P.J.; Jayadev, S.; Davis, M.; Hoffer, Z.S.; Montine, T.J.; Gonzalez-Cuyar, L.F.; Bird, T.D.; Keene, C.D. Neuropathological comparison of adult onset and juvenile huntington’s disease with cerebellar atrophy: A report of a father and son. J. Huntingt. Dis. 2017, 6, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Reetz, K.; Costa, A.S.; Mirzazade, S.; Lehmann, A.; Juzek, A.; Rakowicz, M.; Boguslawska, R.; Schöls, L.; Linnemann, C.; Mariotti, C.; et al. Genotype-Specific Patterns of Atrophy Progression Are More Sensitive than Clinical Decline in SCA1, SCA3 and SCA6. Brain 2013, 136, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, E.; Tremblay, L.; Féger, J.; Carras, P.L.; Strick, P.L. The Cerebellum Communicates with the Basal Ganglia. Nat. Neurosci. 2005, 8, 1491–1493. [Google Scholar] [CrossRef]
- Bostan, A.C.; Dum, R.P.; Strick, P.L. The Basal Ganglia Communicate with the Cerebellum. Proc. Natl. Acad. Sci. USA 2010, 107, 8452–8456. [Google Scholar] [CrossRef]
- Bostan, A.C.; Strick, P.L. The Basal Ganglia and the Cerebellum: Nodes in an Integrated Network. Nat. Rev. Neurosci. 2018, 19, 338–350. [Google Scholar] [CrossRef]
- Washburn, S.; Oñate, M.; Yoshida, J.; Vera, J.; Bhuvanasundaram, R.; Khatami, L.; Nadim, F.; Khodakhah, K. The Cerebellum Directly Modulates the Substantia Nigra Dopaminergic Activity. Nat. Neurosci. 2024, 27, 497–513. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoh1 | Pax6 | Penk+ Glutamatergic Progenitors, GC Precursors and GCs | |
|---|---|---|---|
| Total cells | 10,706 | 12,538 | 169 |
| Penk | 187 | 162 | N/A |
| Penk % of cells | 1.75 | 1.29 | N/A |
| Ntrk2 | 1352 | 2686 | 25 |
| Ntrk2 % of cells | 12.63 | 21.42 | 14.79 |
| GCs | Golgi Cells | PCs | |
|---|---|---|---|
| Total cells | 477,176 | 3989 | 16,634 |
| Penk scaled expression | 0.01 | 3.85 | 0.02 |
| Penk % of cells | 1.03 | 81.35 | 2.03 |
| Ntrk2 scaled expression | 1.48 | 11.17 | 0.4 |
| Ntrk2 % of cells | 69.47 | 99 | 26.72 |
| Test | Genotype | Age (in Months) | n | Median | Max | Min | Mann-Whitney U | Adjusted p |
|---|---|---|---|---|---|---|---|---|
| Hindpaw clasping | Control | 3 | 9, all male | 0 | 0 | 0 | 40.50 | >0.999 |
| Mutant | 3 | 9, all male | 0 | 0 | 0 | |||
| Control | 8 | 3, 1 male and 2 females | 0 | 0 | 0 | 6.00 | >0.999 | |
| Mutant | 8 | 4, 2 males and 2 females | 0 | 0 | 0 | |||
| Kyphosis test | Control | 3 | 10, 3 males and 7 females | 0 | 0 | 0 | 45.00 | >0.999 |
| Mutant | 3 | 9, 4 males and 5 females | 0 | 0 | 0 | |||
| Control | 8 | 6, 1 male and 5 females | 0 | 0 | 0 | 33.00 | >0.999 | |
| Mutant | 8 | 12, 4 males and 8 females | 0 | 2 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eliseeva, E.; Malik, M.Y.; Minichiello, L. Ablation of TrkB from Enkephalinergic Precursor-Derived Cerebellar Granule Cells Generates Ataxia. Biology 2024, 13, 637. https://doi.org/10.3390/biology13080637
Eliseeva E, Malik MY, Minichiello L. Ablation of TrkB from Enkephalinergic Precursor-Derived Cerebellar Granule Cells Generates Ataxia. Biology. 2024; 13(8):637. https://doi.org/10.3390/biology13080637
Chicago/Turabian StyleEliseeva, Elena, Mohd Yaseen Malik, and Liliana Minichiello. 2024. "Ablation of TrkB from Enkephalinergic Precursor-Derived Cerebellar Granule Cells Generates Ataxia" Biology 13, no. 8: 637. https://doi.org/10.3390/biology13080637
APA StyleEliseeva, E., Malik, M. Y., & Minichiello, L. (2024). Ablation of TrkB from Enkephalinergic Precursor-Derived Cerebellar Granule Cells Generates Ataxia. Biology, 13(8), 637. https://doi.org/10.3390/biology13080637