

Pathological Role of High Sugar in Mitochondrial Respiratory Chain Defect-Augmented Mitochondrial Stress

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. NARP Cells (Cybrids) and Parental 143B Cells
2.2. Measurement of Cell Viability Using the MTT Assay
2.3. Preparation of the Cells for Imaging
2.4. Chemical and Fluorescent Dye Loading for Fluorescence Measurement of Mitochondrial Events
2.5. Imaging Analysis of Living Cells
2.6. Statistical Analysis
3. Results
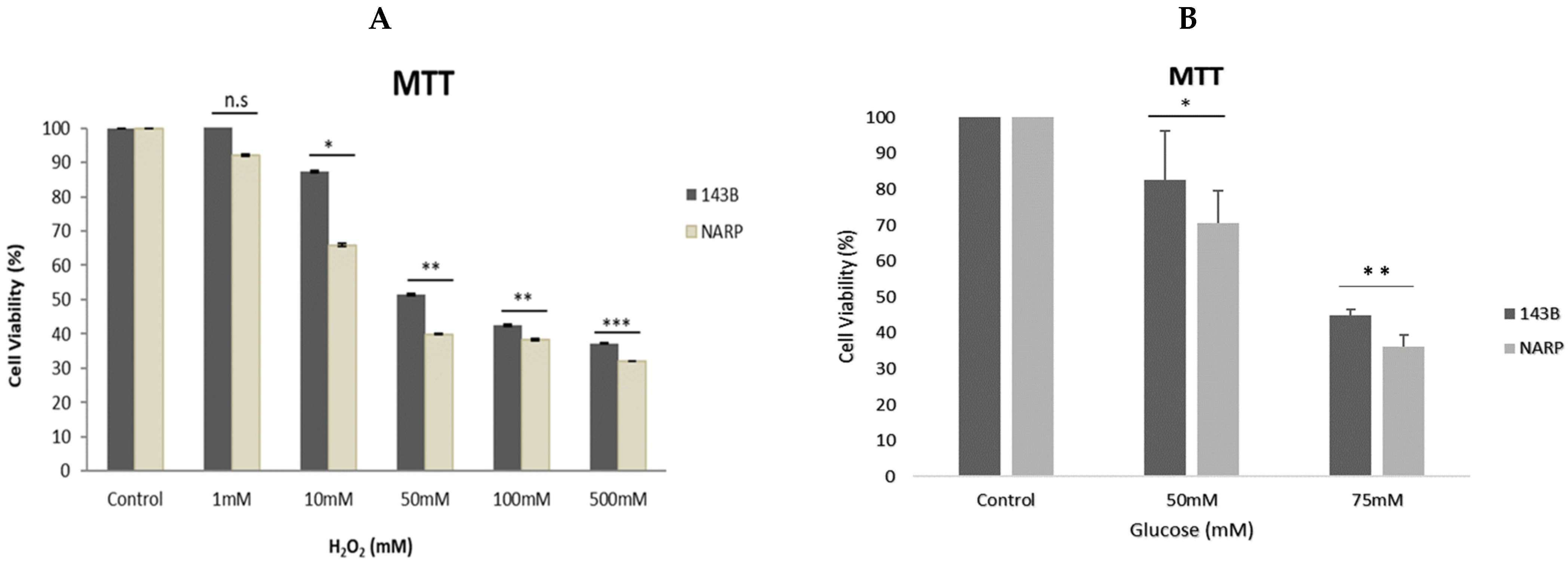
3.1. Assessing Cell Viability under Different Stress Conditions
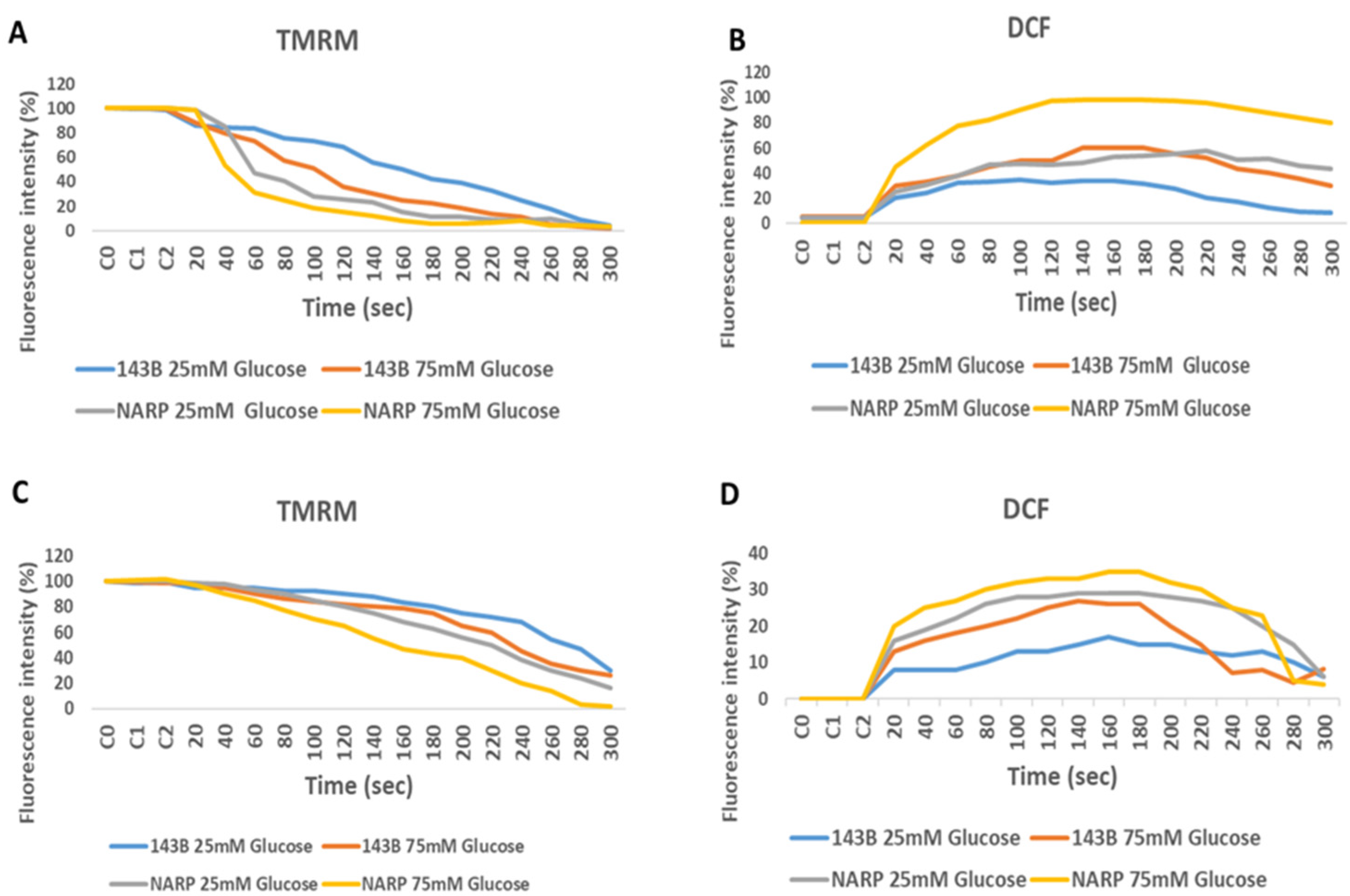
3.2. Determining the Toxicity of High Glucose and Mitochondrial Calcium [Ca2+]m Overload on 143B Cells and NARP Cybrids
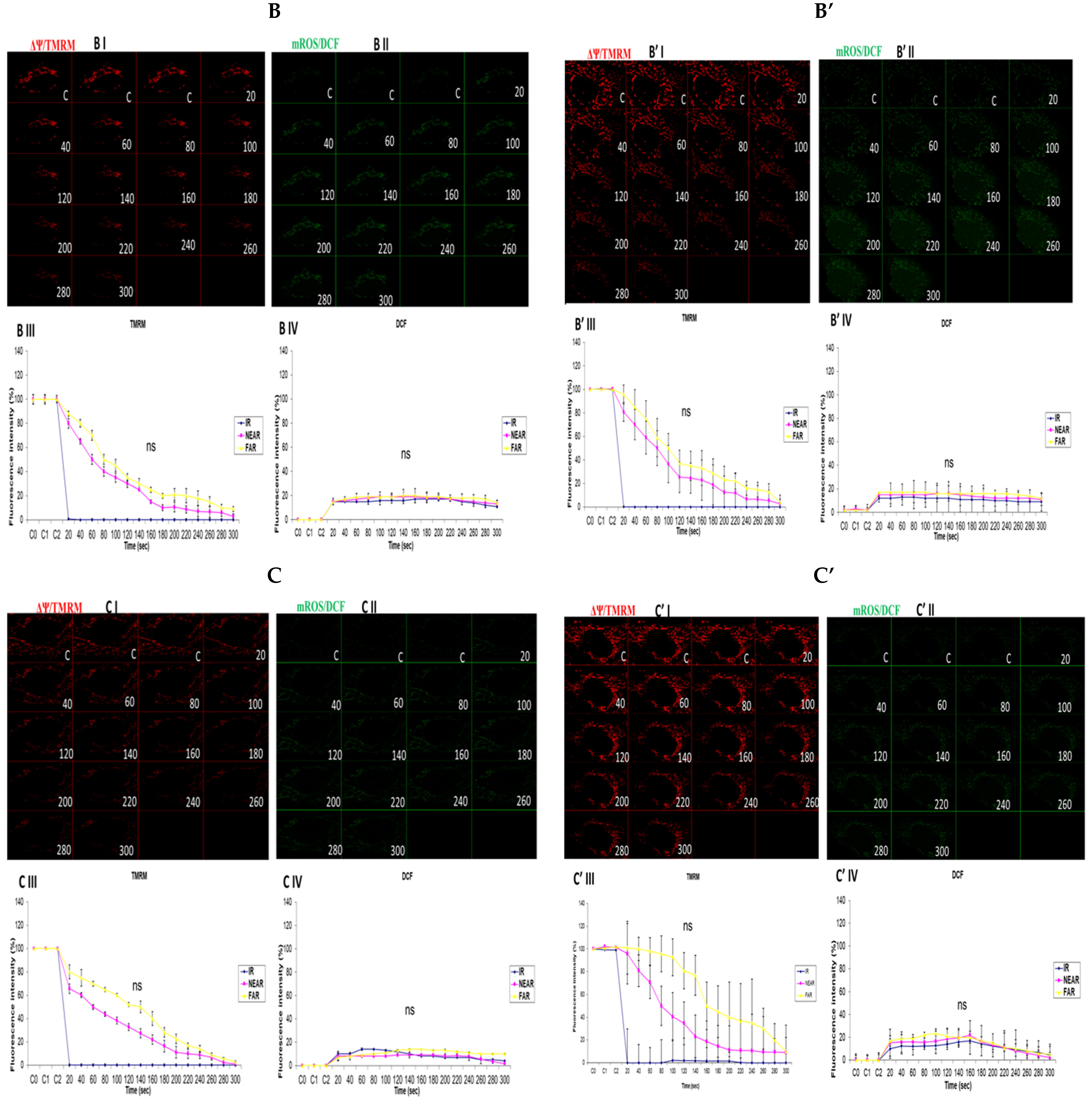
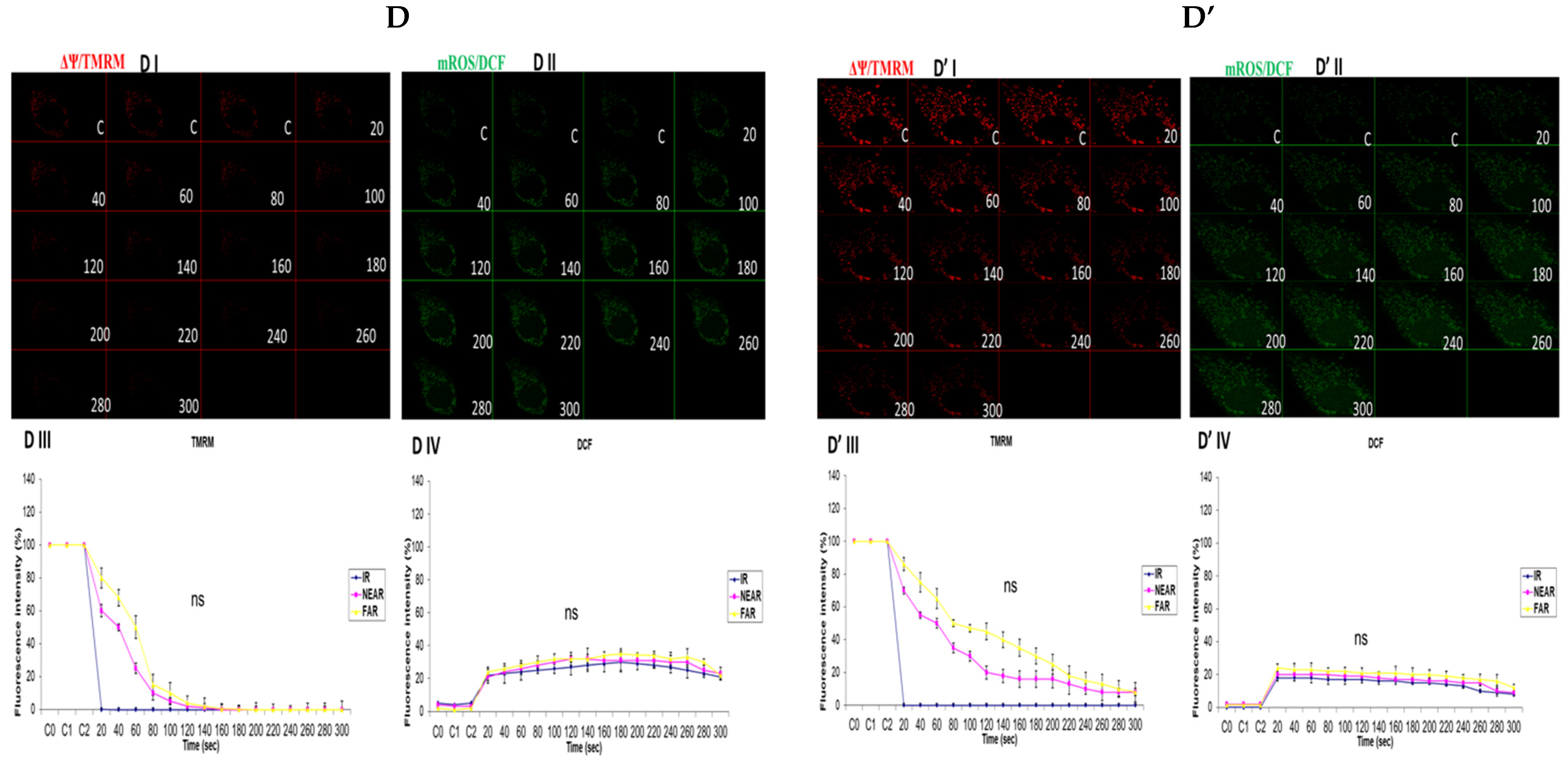
3.3. Depicting How Mitochondrial Calcium Overload [Ca2+]m Modulates High Glucose Toxicity on Respiratory Chain Defect-Augmented Mitochondrial Reactive Oxygen Species (ROS) Production and Membrane Potential (ΔΨ) Depolarization in Parental 143B Cells and NARP Cybrids
3.4. Depicting How Mitochondrial Calcium Overload [Ca2+]m Modulates High Glucose Toxicity on Respiratory Chain Defect-Augmented Mitochondrial Reactive Oxygen Species (ROS) Production and Membrane Potential (ΔΨ) Depolarization in Parental 143B Cells and NARP Cybrids under Antioxidant Conditions
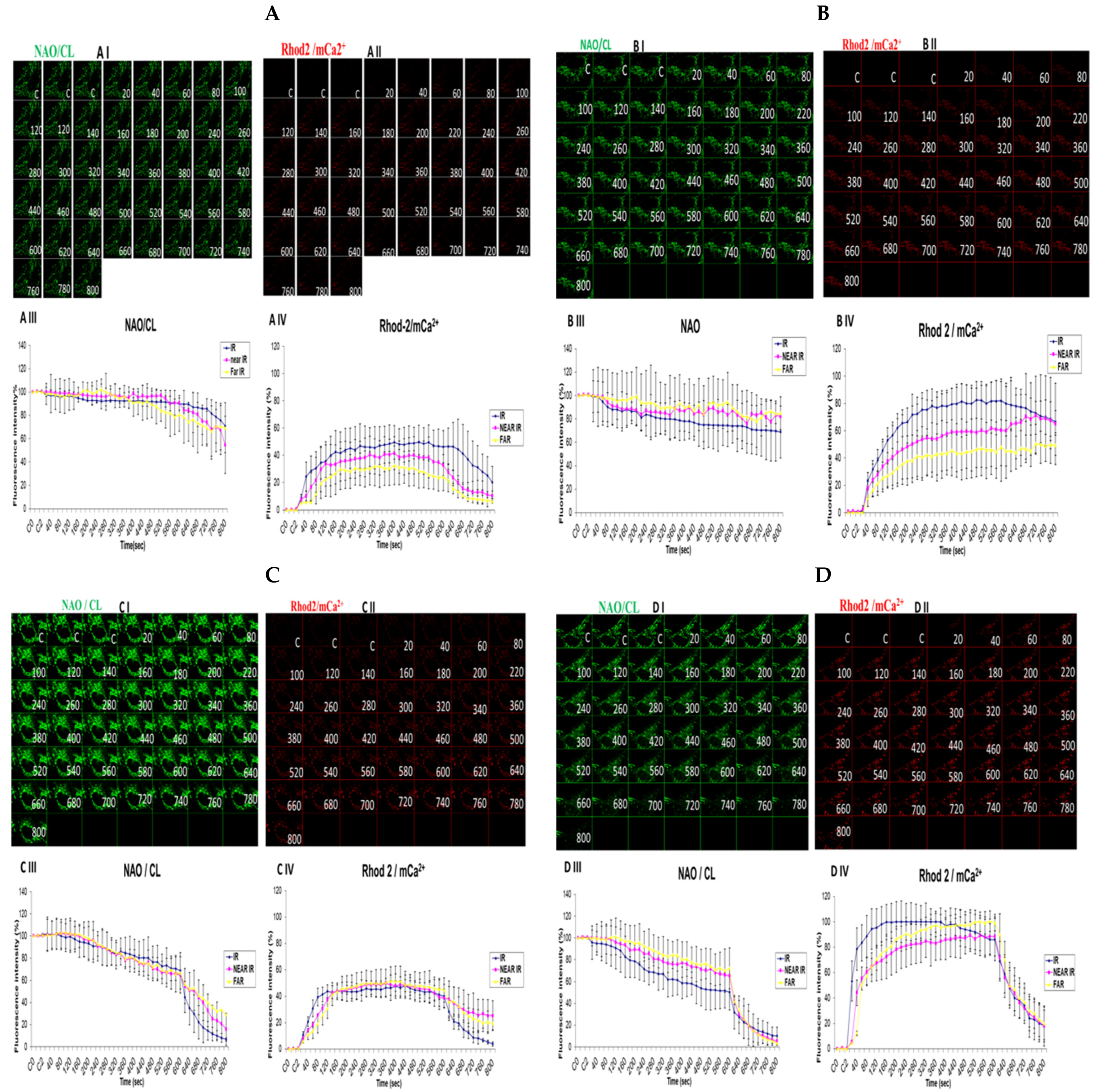
3.5. Demonstrating How Mitochondrial Calcium Overload Modulates High Glucose Toxicity on Respiratory Chain Defect-Augmented Mitochondrial Cardiolipin (CL) Remodeling and Calcium (Ca2+) Homeostasis in Parental 143B Cells and NARP Cybrids under Ca2+-Treated Hepex Conditions
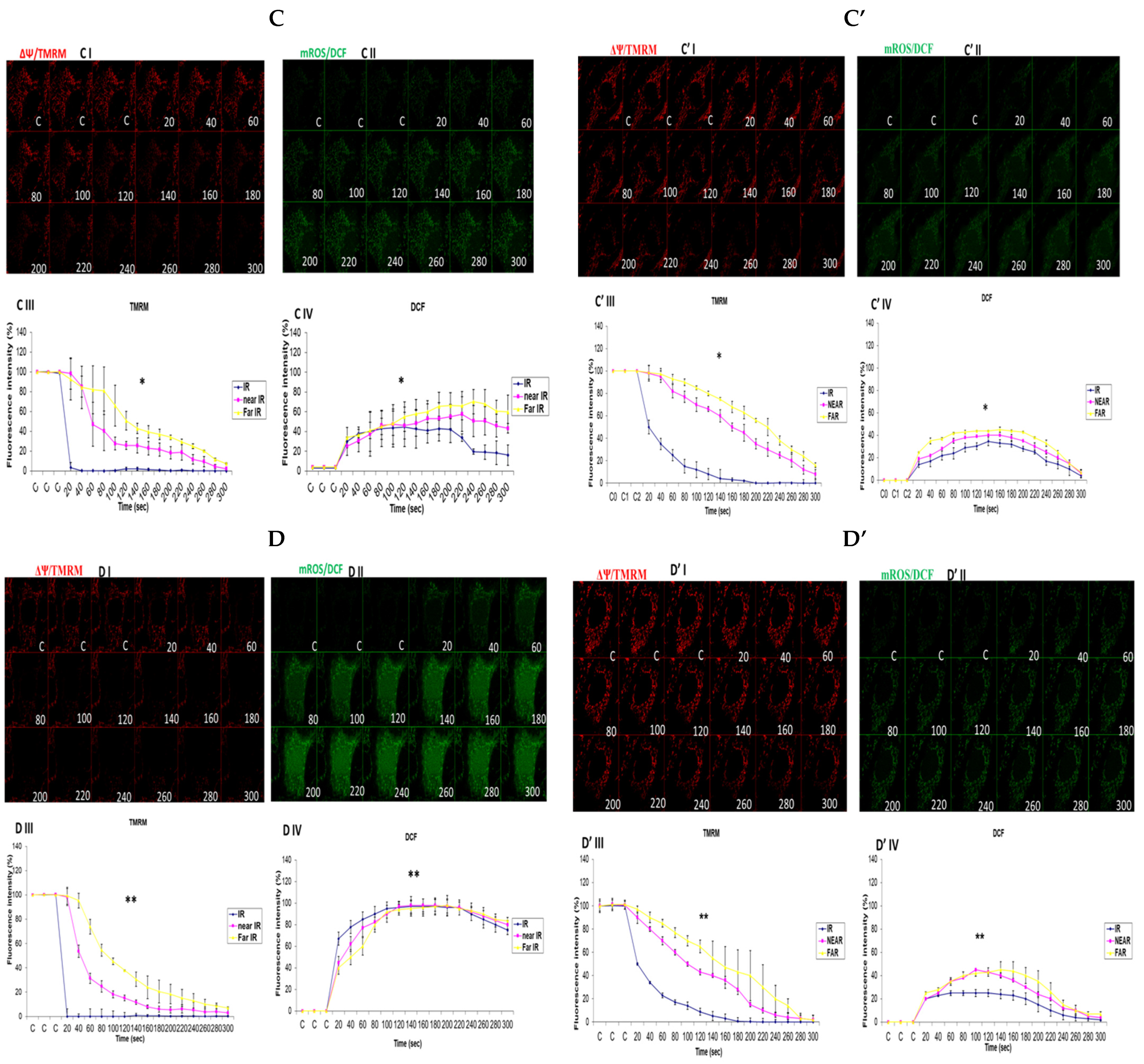
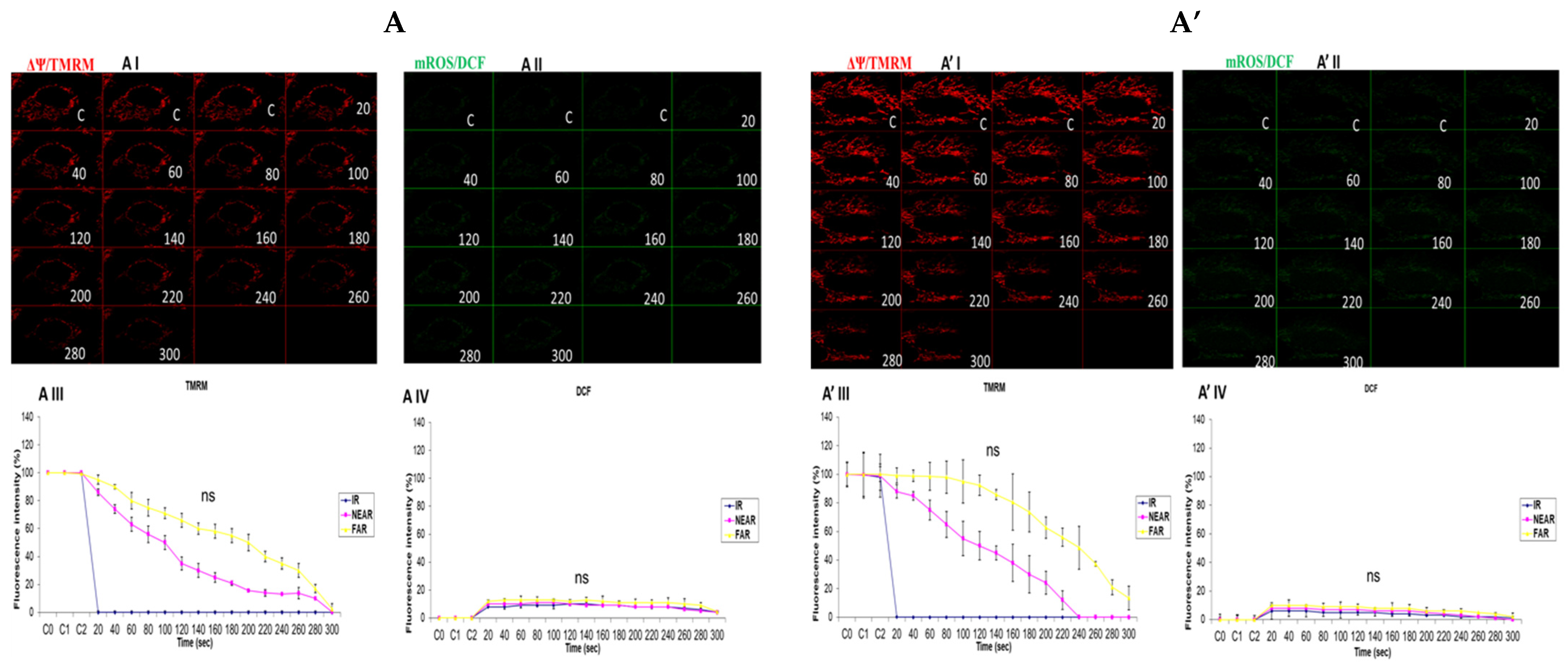
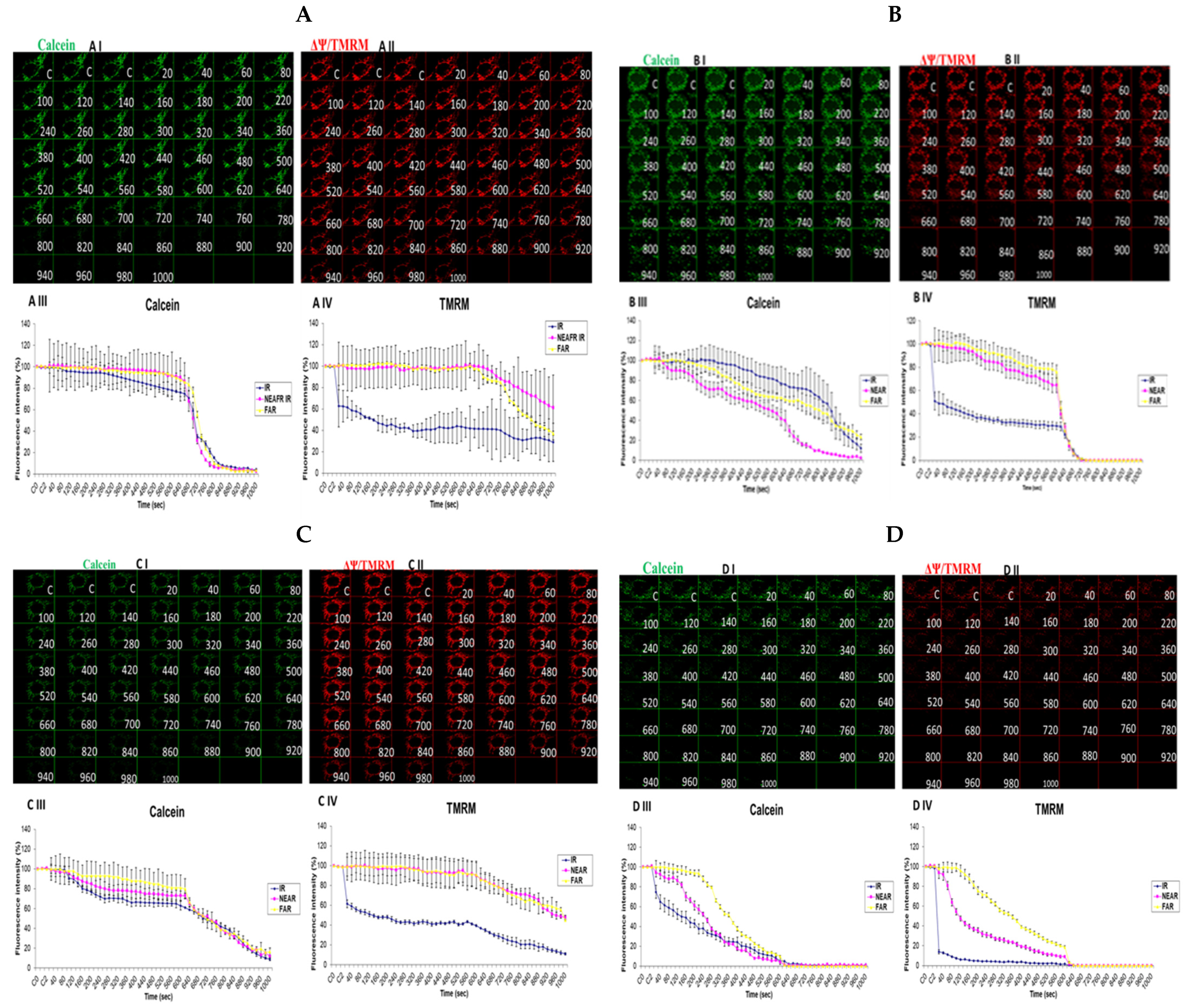
3.6. Investigation of How High Glucose Augments Respiratory Chain Defect-Augmented Mitochondrial Permeability Transition Pore (MPTP), Including Transient (t-MPT) and Permanent (p-MPT), on Parental 143B Cells and NARP Cybrids
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Green, K.; Brand, M.D.; Murphy, M.P. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004, 53 (Suppl. S1), S110–S118. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet. 2005, 14 (Suppl. S2), R283–R289. [Google Scholar] [CrossRef]
- Vanhorebeek, I.; De Vos, R.; Mesotten, D.; Wouters, P.J.; De Wolf-Peeters, C.; Van den Berghe, G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 2005, 365, 53–59. [Google Scholar] [CrossRef]
- Kemppainen, E.; George, J.; Garipler, G.; Tuomela, T.; Kiviranta, E.; Soga, T.; Dunn, C.D.; Jacobs, H.T. Correction: Mitochondrial Dysfunction Plus High-Sugar Diet Provokes a Metabolic Crisis That Inhibits Growth. PLoS ONE 2016, 11, e0151421. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef]
- Okoh, V.I.; Campos, H.M.; Yasmin de Oliveira Ferreira, P.; Pereira, R.M.; Souza Silva, Y.; Arruda, E.L.; Pagliarani, B.; de Almeida Ribeiro Oliveira, G.; Liao, L.M.; Franco Dos Santos, G.; et al. Chrysin bonded to beta-d-glucose tetraacetate enhances its protective effects against the neurotoxicity induced by aluminum in Swiss mice. J. Pharm. Pharmacol. 2024, 76, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288. [Google Scholar] [CrossRef]
- Lange, C.; Nett, J.H.; Trumpower, B.L.; Hunte, C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 2001, 20, 6591–6600. [Google Scholar] [CrossRef]
- Koshkin, V.; Greenberg, M.L. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem. J. 2002, 364, 317–322. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schagger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef]
- Yu, T.; Jhun, B.S.; Yoon, Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid. Redox Signal 2011, 14, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Baracca, A.; Barogi, S.; Carelli, V.; Lenaz, G.; Solaini, G. Catalytic activities of mitochondrial ATP synthase in patients with mitochondrial DNA T8993G mutation in the ATPase 6 gene encoding subunit a. J. Biol. Chem. 2000, 275, 4177–4182. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Han, J. Mitochondria-Targeted Antioxidants for the Treatment of Cardiovascular Disorders. Adv. Exp. Med. Biol. 2017, 982, 621–646. [Google Scholar] [CrossRef]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef]
- Huser, J.; Rechenmacher, C.E.; Blatter, L.A. Imaging the permeability pore transition in single mitochondria. Biophys. J. 1998, 74, 2129–2137. [Google Scholar] [CrossRef]
- D’Aurelio, M.; Vives-Bauza, C.; Davidson, M.M.; Manfredi, G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Hum. Mol. Genet. 2010, 19, 374–386. [Google Scholar] [CrossRef]
- Carrozzo, R.; Tessa, A.; Vazquez-Memije, M.E.; Piemonte, F.; Patrono, C.; Malandrini, A.; Dionisi-Vici, C.; Vilarinho, L.; Villanova, M.; Schagger, H.; et al. The T9176G mtDNA mutation severely affects ATP production and results in Leigh syndrome. Neurology 2001, 56, 687–690. [Google Scholar] [CrossRef] [PubMed]
- De Meirleir, L.; Seneca, S.; Lissens, W.; De Clercq, I.; Eyskens, F.; Gerlo, E.; Smet, J.; Van Coster, R. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J. Med. Genet. 2004, 41, 120–124. [Google Scholar] [CrossRef]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137–4142. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in Mitochondrial Oxidative Stress and Mitophagy in Subjects with Prediabetes and Type 2 Diabetes Mellitus. Front. Endocrinol. 2017, 8, 347. [Google Scholar] [CrossRef]
- Jou, M.J.; Peng, T.I.; Reiter, R.J. Protective stabilization of mitochondrial permeability transition and mitochondrial oxidation during mitochondrial Ca2+ stress by melatonin’s cascade metabolites C3-OHM and AFMK in RBA1 astrocytes. J. Pineal Res. 2019, 66, e12538. [Google Scholar] [CrossRef]
- Jou, M.J.; Peng, T.I.; Yu, P.Z.; Jou, S.B.; Reiter, R.J.; Chen, J.Y.; Wu, H.Y.; Chen, C.C.; Hsu, L.F. Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J. Pineal Res. 2007, 43, 389–403. [Google Scholar] [CrossRef]
- Cai, D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol. Metab. 2013, 24, 40–47. [Google Scholar] [CrossRef]
- Huang, W.Y.; Jou, M.J.; Peng, T.I. mtDNA T8993G mutation-induced F1F0-ATP synthase defect augments mitochondrial dysfunction associated with hypoxia/reoxygenation: The protective role of melatonin. PLoS ONE 2013, 8, e81546. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Slabe, T.J.; Stoll, M.S.; Minkler, P.E.; Hassan, M.O.; Tandler, B.; Hoppel, C.L. Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: Role of cardiolipin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H258–H267. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Serena, D.; Ruggiero, F.M. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic. Biol. Med. 1999, 27, 42–50. [Google Scholar] [CrossRef]
- Petrosillo, G.; Ruggiero, F.M.; Di Venosa, N.; Paradies, G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: Role of reactive oxygen species and cardiolipin. FASEB J. 2003, 17, 714–716. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: Induction of permeability transition and cytochrome c release. FEBS Lett. 2006, 580, 6311–6316. [Google Scholar] [CrossRef]
- Petrosillo, G.; Moro, N.; Ruggiero, F.M.; Paradies, G. Melatonin inhibits cardiolipin peroxidation in mitochondria and prevents the mitochondrial permeability transition and cytochrome c release. Free Radic. Biol. Med. 2009, 47, 969–974. [Google Scholar] [CrossRef]
- Murphy, M.P. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997, 15, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Smith, R.A. Drug delivery to mitochondria: The key to mitochondrial medicine. Adv. Drug Deliv. Rev. 2000, 41, 235–250. [Google Scholar] [CrossRef]
- Park, J.T.; Lee, Y.S.; Cho, K.A.; Park, S.C. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res. Rev. 2018, 47, 176–182. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Fatima, M.T.; Bhat, A.A.; Nisar, S.; Fakhro, K.A.; Al-Shabeeb Akil, A.S. The role of dietary antioxidants in type 2 diabetes and neurodegenerative disorders: An assessment of the benefit profile. Heliyon 2023, 9, e12698. [Google Scholar] [CrossRef] [PubMed]
- Tuell, D.S.; Los, E.A.; Ford, G.A.; Stone, W.L. The Role of Natural Antioxidant Products That Optimize Redox Status in the Prevention and Management of Type 2 Diabetes. Antioxidants 2023, 12, 1139. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cham, E.D.; Peng, T.-I.; Jou, M.-J. Pathological Role of High Sugar in Mitochondrial Respiratory Chain Defect-Augmented Mitochondrial Stress. Biology 2024, 13, 639. https://doi.org/10.3390/biology13080639
Cham ED, Peng T-I, Jou M-J. Pathological Role of High Sugar in Mitochondrial Respiratory Chain Defect-Augmented Mitochondrial Stress. Biology. 2024; 13(8):639. https://doi.org/10.3390/biology13080639
Chicago/Turabian StyleCham, Ebrima D., Tsung-I Peng, and Mei-Jie Jou. 2024. "Pathological Role of High Sugar in Mitochondrial Respiratory Chain Defect-Augmented Mitochondrial Stress" Biology 13, no. 8: 639. https://doi.org/10.3390/biology13080639
APA StyleCham, E. D., Peng, T.-I., & Jou, M.-J. (2024). Pathological Role of High Sugar in Mitochondrial Respiratory Chain Defect-Augmented Mitochondrial Stress. Biology, 13(8), 639. https://doi.org/10.3390/biology13080639