
Metrics of Genomic Complexity in the Evolution of Bacterial Endosymbiosis

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Set and Species Phylogeny
2.2. Genomic Metrics and Parameter Calculation
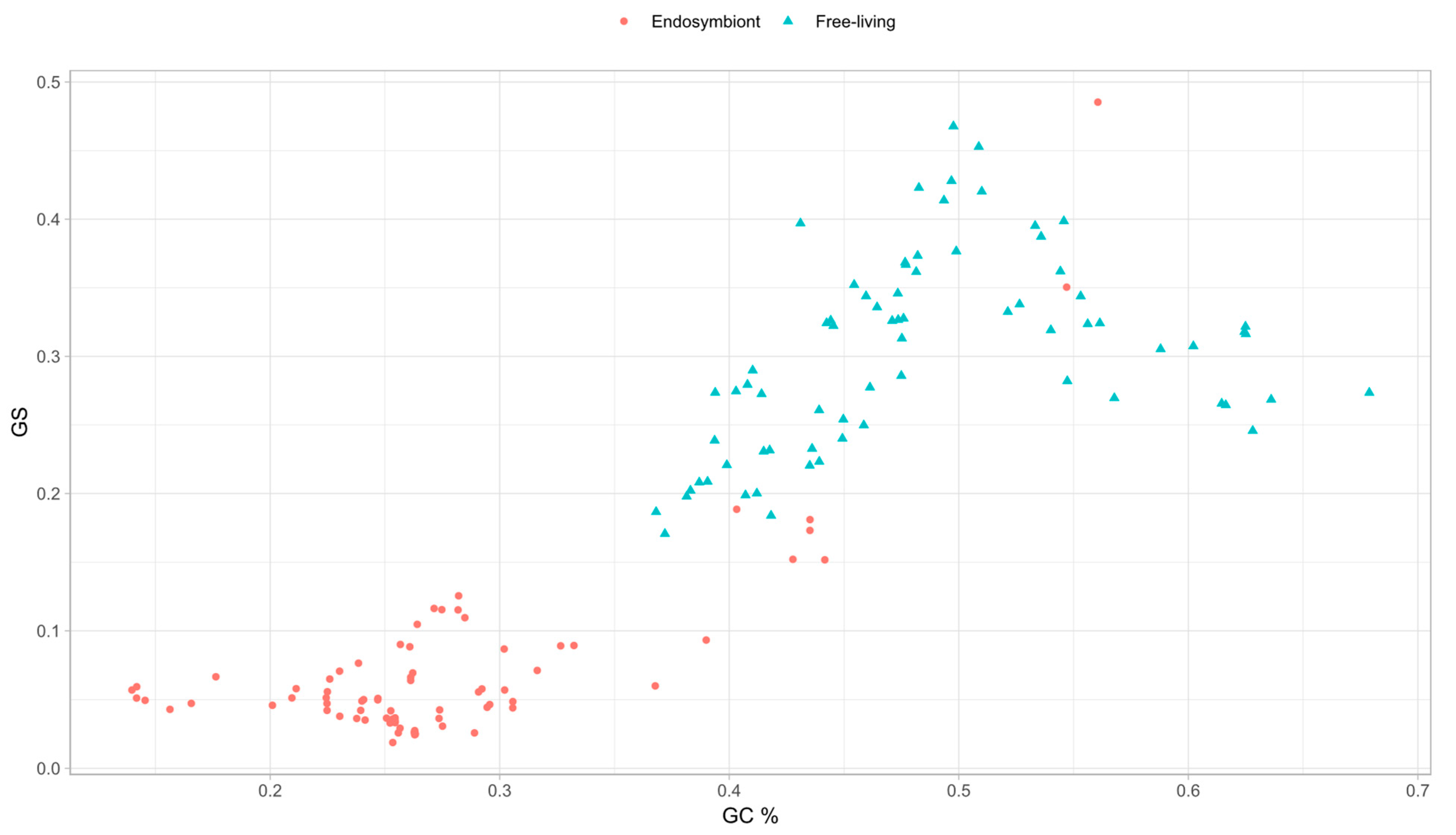
2.3. Genome Signature (GS)
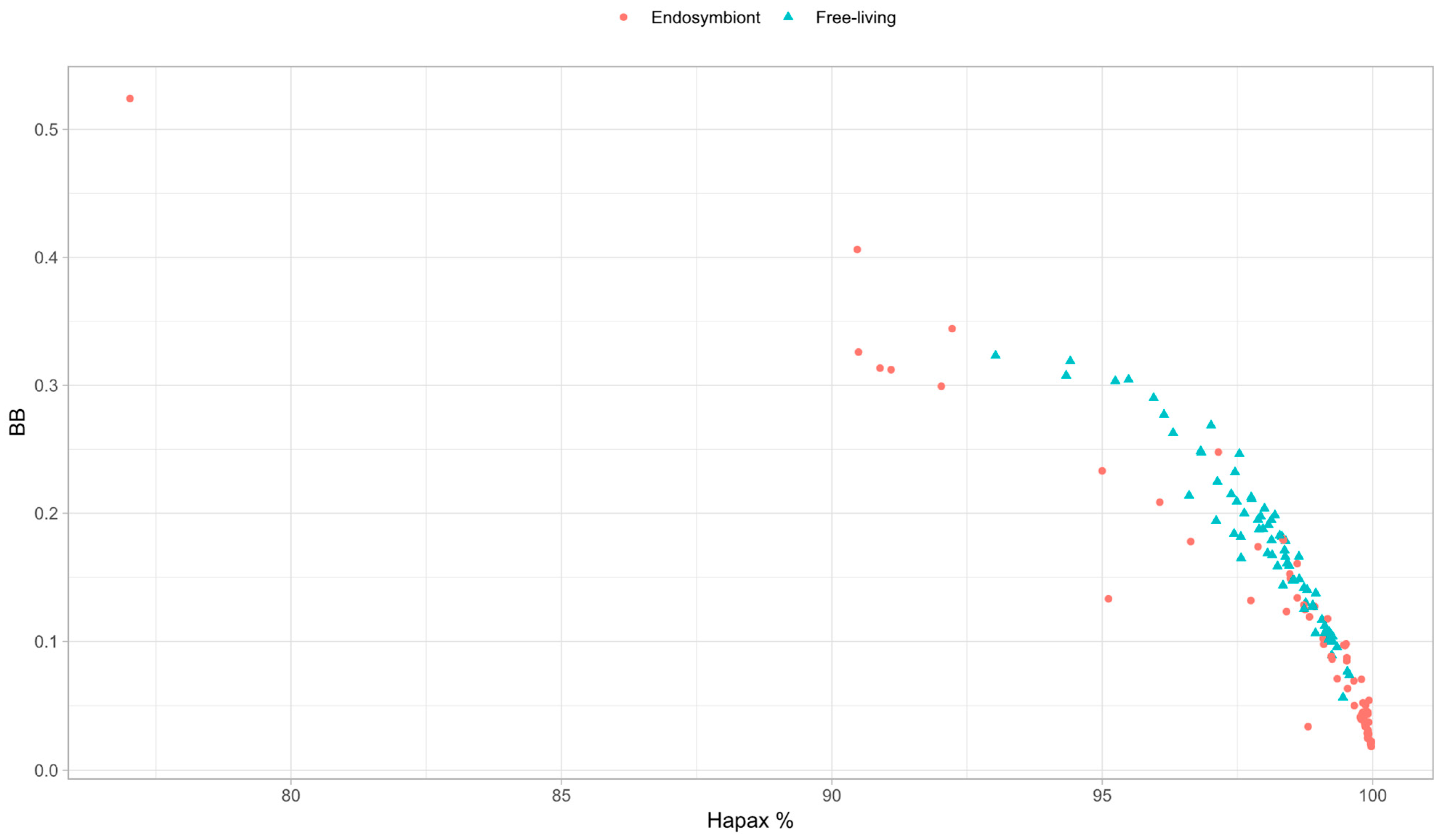
2.4. Biobit (BB)
2.5. Statistical Analyses
2.6. Phylogenetic Signal
3. Results and Discussion
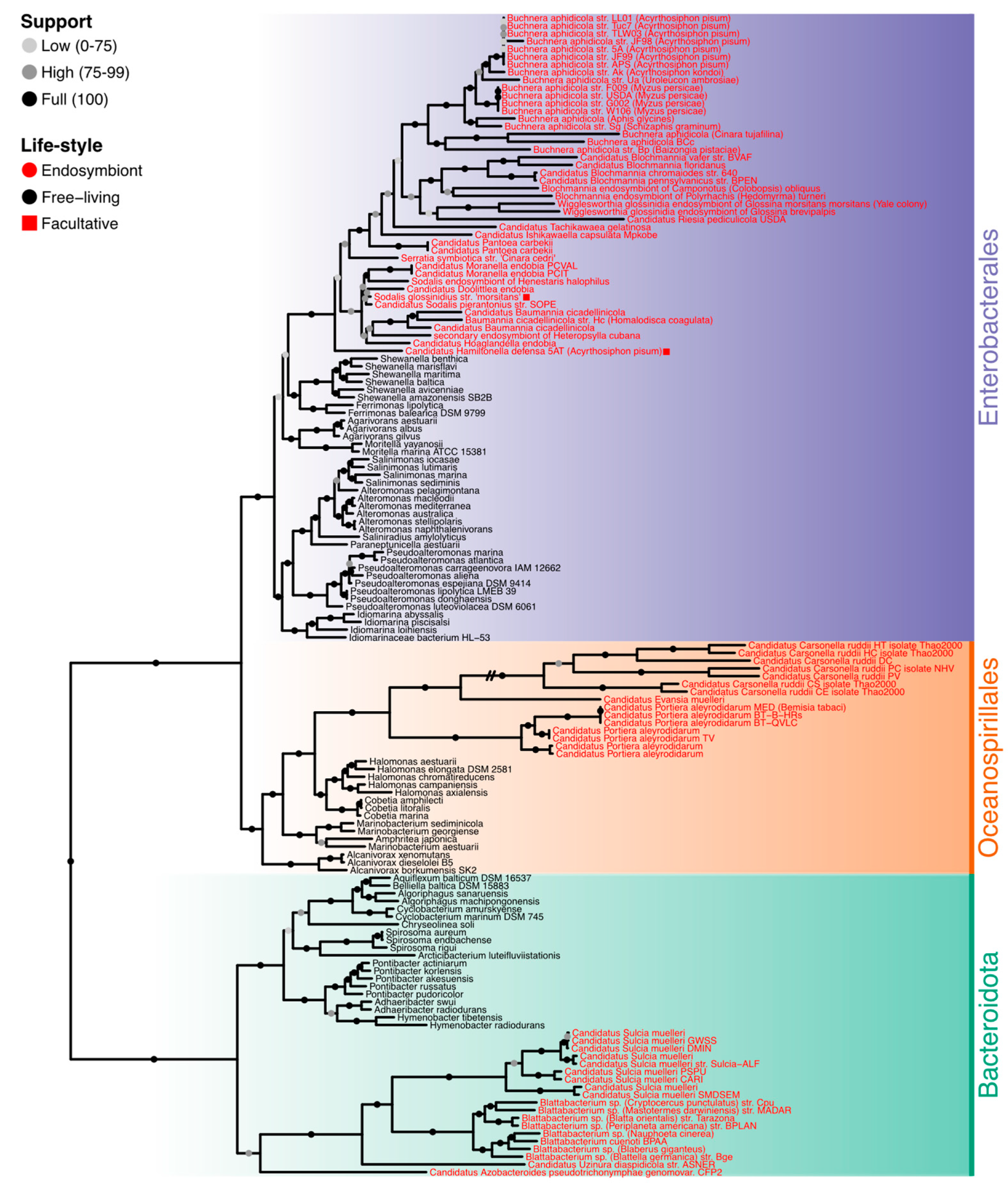
3.1. Phylogenetic Analyses
3.2. Phylogenetic Signal
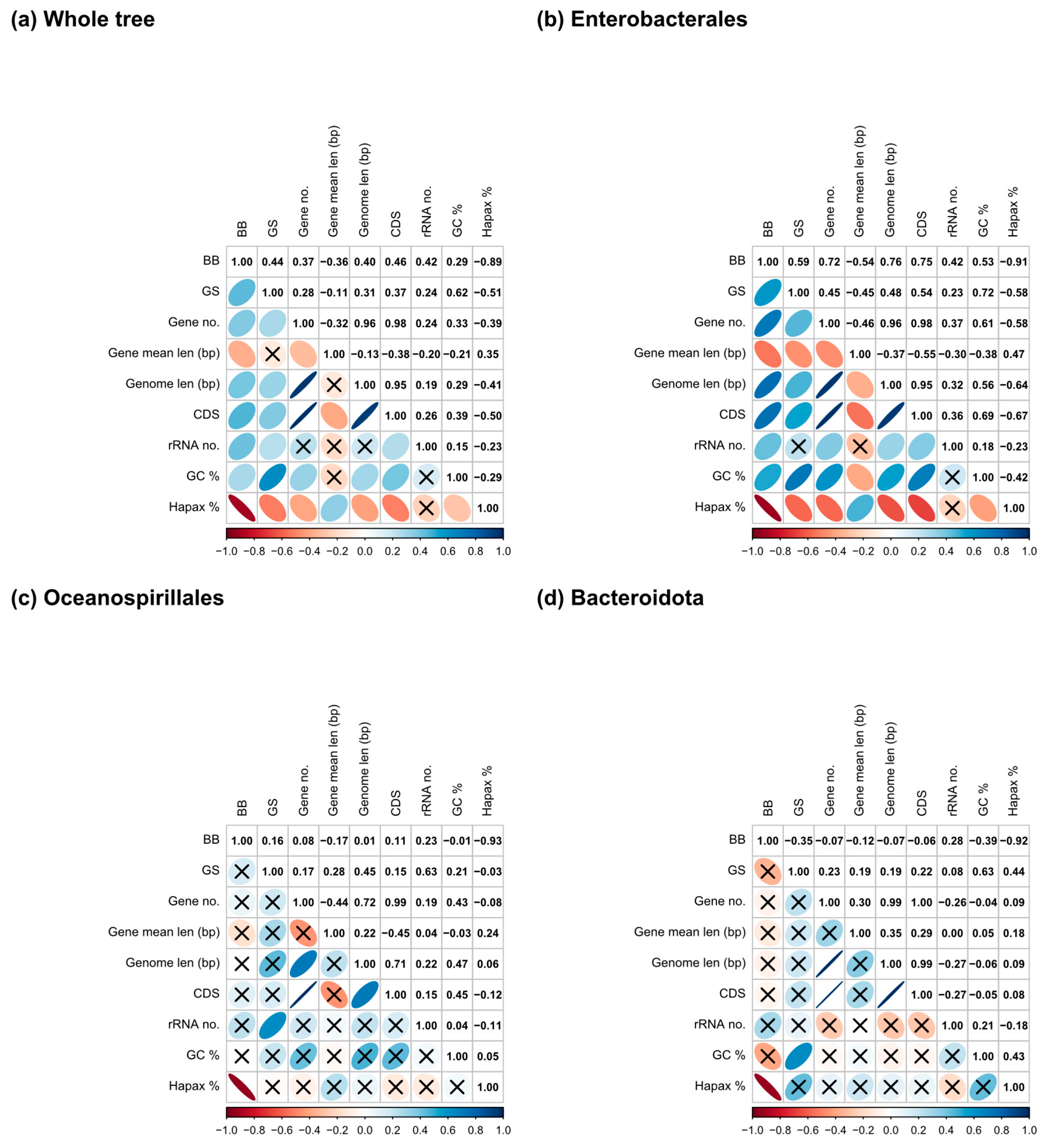
3.3. Metrics, Genome Parameters, and Phylogenetic Correlations
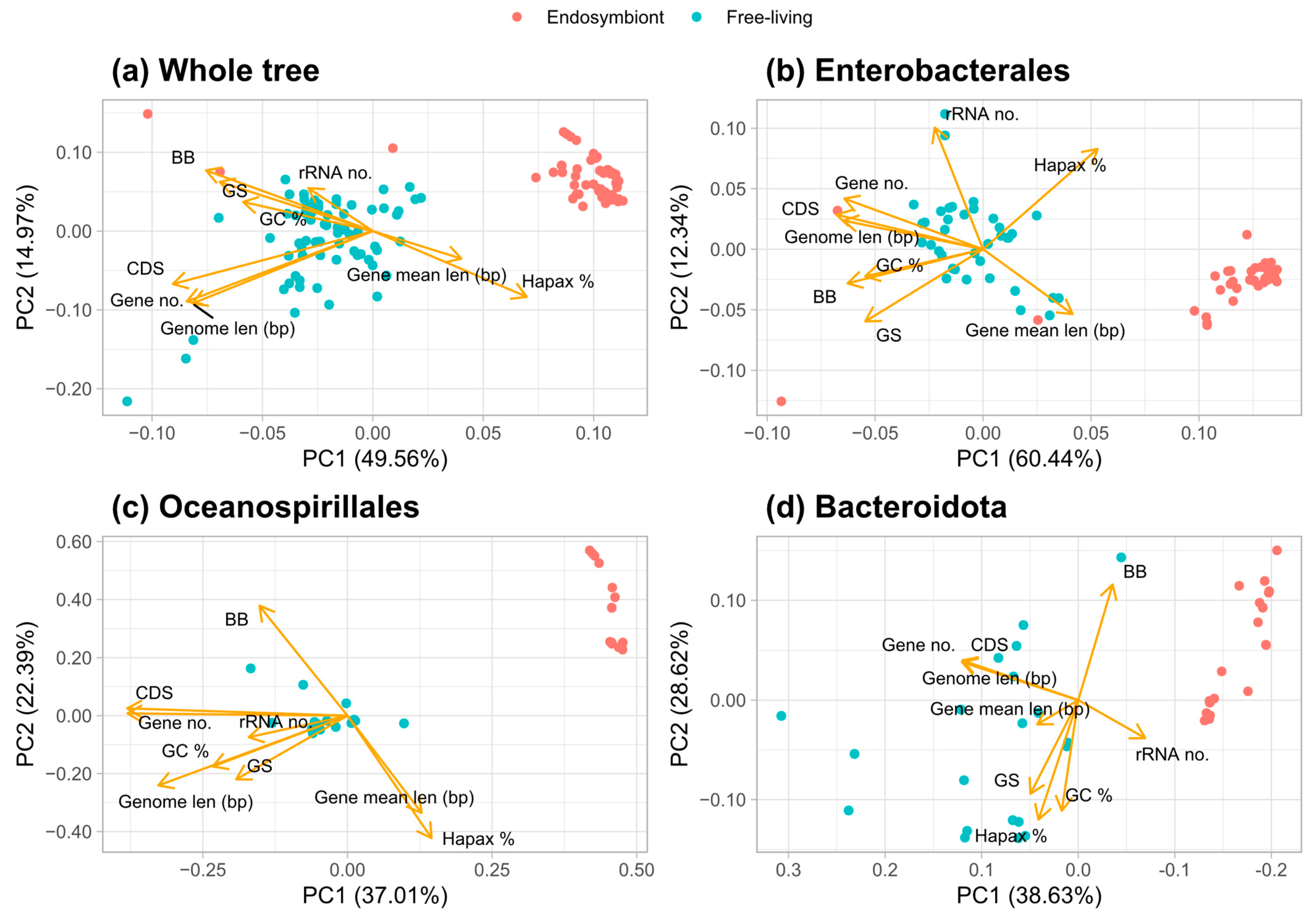
3.4. Principal Component Analysis of Traits That Discriminate Between Bacteria Lifestyles
3.5. Genomic Base Composition Drives the GS and BB Values
3.6. Outlier Analyses of BB
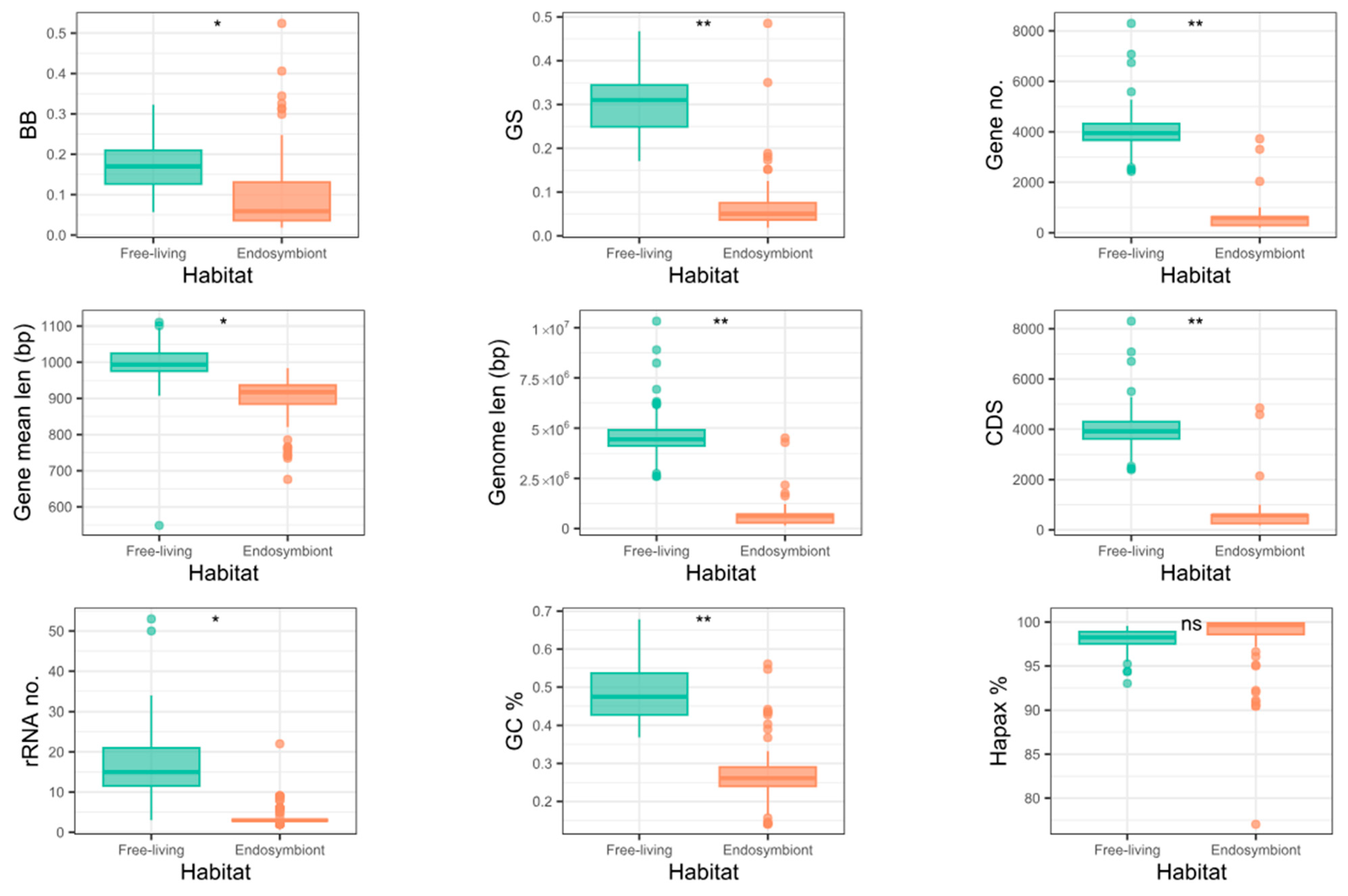
3.7. Genome Complexity and Metrics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adami, C. What Is Complexity? Bioessays 2002, 24, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- McShea, D.W.; Brandon, R.N. Biology’s First Law: The Tendency for Diversity and Complexity to Increase in Evolutionary Systems, 1st ed.; University of Chicago Press: Chicago, IL, USA, 2010. [Google Scholar]
- Koonin, E.V. The Meaning of Biological Information. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150065. [Google Scholar] [CrossRef]
- Heim, N.A.; Payne, J.L.; Finnegan, S.; Knope, M.L.; Kowalewski, M.; Lyons, S.K.; McShea, D.W.; Novack-Gottshall, P.M.; Smith, F.A.; Wang, S.C. Hierarchical Complexity and the Size Limits of Life. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171039. [Google Scholar] [CrossRef] [PubMed]
- Adami, C.; Cerf, N.J.; Kellogg, W.K. Physical Complexity of Symbolic Sequences. Physica D Nonlinear Phenom. 2000, 137, 62–69. [Google Scholar]
- Adami, C.; Ofria, C.; Collier, T.C. Evolution of Biological Complexity. Proc. Natl. Acad. Sci. USA 2000, 97, 4463–4468. [Google Scholar] [CrossRef]
- Adami, C. What Is Information? Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150230. [Google Scholar] [CrossRef]
- Moya, A. The Calculus of Life, 1st ed.; Springer Briefs in Biology; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar] [CrossRef]
- Moya, A.; Oliver, J.L.; Verdú, M.; Delaye, L.; Arnau, V.; Bernaola-Galván, P.; de la Fuente, R.; Díaz, W.; Gómez-Martín, C.; González, F.M.; et al. Driven Progressive Evolution of Genome Sequence Complexity in Cyanobacteria. Sci. Rep. 2020, 10, 19073. [Google Scholar] [CrossRef]
- Adami, C. The Evolution of Biological Information: How Evolution Creates Complexity, from Viruses to Brains, 1st ed.; Princeton University Press: Princeton, NJ, USA, 2024. [Google Scholar]
- De la Fuente, R.; Díaz-Villanueva, W.; Arnau, V.; Moya, A. Genomic Signature in Evolutionary Biology: A Review. Biology 2023, 12, 322. [Google Scholar] [CrossRef]
- Bonnici, V.; Manca, V. Informational Laws of Genome Structures. Sci. Rep. 2016, 6, 28840. [Google Scholar] [CrossRef]
- Delaye, L.; Moya, A. Evolution of Reduced Prokaryotic Genomes and the Minimal Cell Concept: Variations on a Theme. BioEssays 2010, 32, 281–287. [Google Scholar] [CrossRef]
- Moran, N.A.; Bennett, G.M. The Tiniest Tiny Genomes. Annu. Rev. Microbiol. 2014, 68, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Husnik, F.; Tashyreva, D.; Boscaro, V.; George, E.E.; Lukeš, J.; Keeling, P.J. Bacterial and Archaeal Symbioses with Protists. Curr. Biol. 2021, 31, R862–R877. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An Ongoing Census of Bacterial and Archaeal Diversity Through a Phylogenetically Consistent, Rank Normalized and Complete Genome-Based Taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Pagel, M. Inferring the Historical Patterns of Biological Evolution. Nature 1999, 401, 877–884. [Google Scholar] [CrossRef]
- Revell, L.J. Size-Correction and Principal Components for Interspecific Comparative Studies. Evolution 2009, 63, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Revell, L.J. Phytools: An R Package for Phylogenetic Comparative Biology (and Other Things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Holm, S. A Simple Sequentially Rejective Multiple Test Procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Blomberg, S.P.; Garland, T.; Ives, A.R. Testing for Phylogenetic Signal in Comparative Data: Behavioral Traits Are More Labile. Evolution 2003, 57, 717–745. [Google Scholar] [CrossRef]
- Brandis, G. Reconstructing the Evolutionary History of a Highly Conserved Operon Cluster in Gammaproteobacteria and Bacilli. Genome Biol. Evol. 2021, 13, evab041. [Google Scholar] [CrossRef]
- Gil, R.; Belda, E.; Gosalbes, M.J.; Delaye, L.; Vallier, A.; Vincent-Monégat, C.; Heddi, A.; Silva, F.J.; Moya, A.; Latorre, A. Massive Presence of Insertion Sequences in the Genome of SOPE, the Primary Endosymbiont of the Rice Weevil Sitophilus Oryzae. Int. Microbiol. 2008, 11, 41–48. [Google Scholar] [CrossRef]
- Degnan, P.H.; Yu, Y.; Sisneros, N.; Wing, R.A.; Moran, N.A. Hamiltonella Defensa, Genome Evolution of Protective Bacterial Endosymbiont from Pathogenic Ancestors. Proc. Natl. Acad. Sci. USA 2009, 106, 9063–9068. [Google Scholar] [CrossRef]
- Belda, E.; Moya, A.; Bentley, S.; Silva, F.J. Mobile Genetic Element Proliferation and Gene Inactivation Impact over the Genome Structure and Metabolic Capabilities of Sodalis Glossinidius, the Secondary Endosymbiont of Tsetse Flies. BMC Genom. 2010, 11, 449. [Google Scholar] [CrossRef]
- Song, H.; Hwang, J.; Yi, H.; Ulrich, R.L.; Yu, Y.; Nierman, W.C.; Kim, H.S. The Early Stage of Bacterial Genome-Reductive Evolution in the Host. PLoS Pathog. 2010, 6, e1000922. [Google Scholar] [CrossRef]
- Sloan, D.B.; Moran, N.A. Genome Reduction and Co-Evolution Between the Primary and Secondary Bacterial Symbionts of Psyllids. Mol. Biol. Evol. 2012, 29, 3781–3792. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Román-Escrivá, P.; Bernabeu, M.; Paganin, E.; Díaz-Villanueva, W.; Verdú, M.; Oliver, J.L.; Arnau, V.; Moya, A. Metrics of Genomic Complexity in the Evolution of Bacterial Endosymbiosis. Biology 2025, 14, 338. https://doi.org/10.3390/biology14040338
Román-Escrivá P, Bernabeu M, Paganin E, Díaz-Villanueva W, Verdú M, Oliver JL, Arnau V, Moya A. Metrics of Genomic Complexity in the Evolution of Bacterial Endosymbiosis. Biology. 2025; 14(4):338. https://doi.org/10.3390/biology14040338
Chicago/Turabian StyleRomán-Escrivá, Pablo, Moisès Bernabeu, Eleonora Paganin, Wladimiro Díaz-Villanueva, Miguel Verdú, José L. Oliver, Vicente Arnau, and Andrés Moya. 2025. "Metrics of Genomic Complexity in the Evolution of Bacterial Endosymbiosis" Biology 14, no. 4: 338. https://doi.org/10.3390/biology14040338
APA StyleRomán-Escrivá, P., Bernabeu, M., Paganin, E., Díaz-Villanueva, W., Verdú, M., Oliver, J. L., Arnau, V., & Moya, A. (2025). Metrics of Genomic Complexity in the Evolution of Bacterial Endosymbiosis. Biology, 14(4), 338. https://doi.org/10.3390/biology14040338