
Whole Genome Sequencing of Kodamaea ohmeri SSK and Its Characterization for Degradation of Inhibitors from Lignocellulosic Biomass

 ,
,  , ,
, ,  and
and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Strain
2.2. Culture Medium
2.3. Kinetics of Strain Growth and Inhibitor Detoxification
2.4. Genome DNA Extraction and Sequencing
2.5. Sequencing Data Accusation and Assembly
2.6. Gene Prediction and Annotation
2.7. Statistical Analyses
3. Results
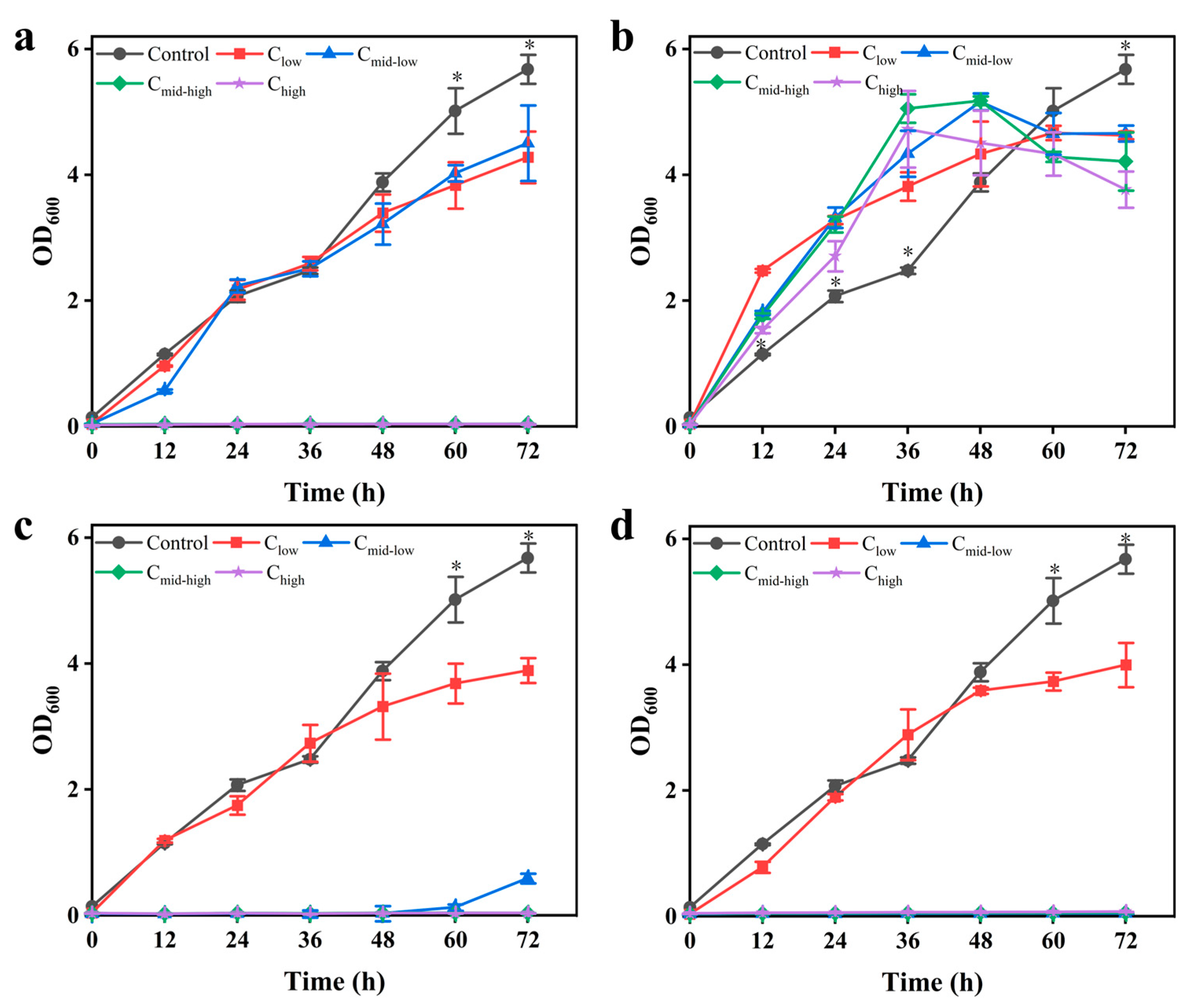
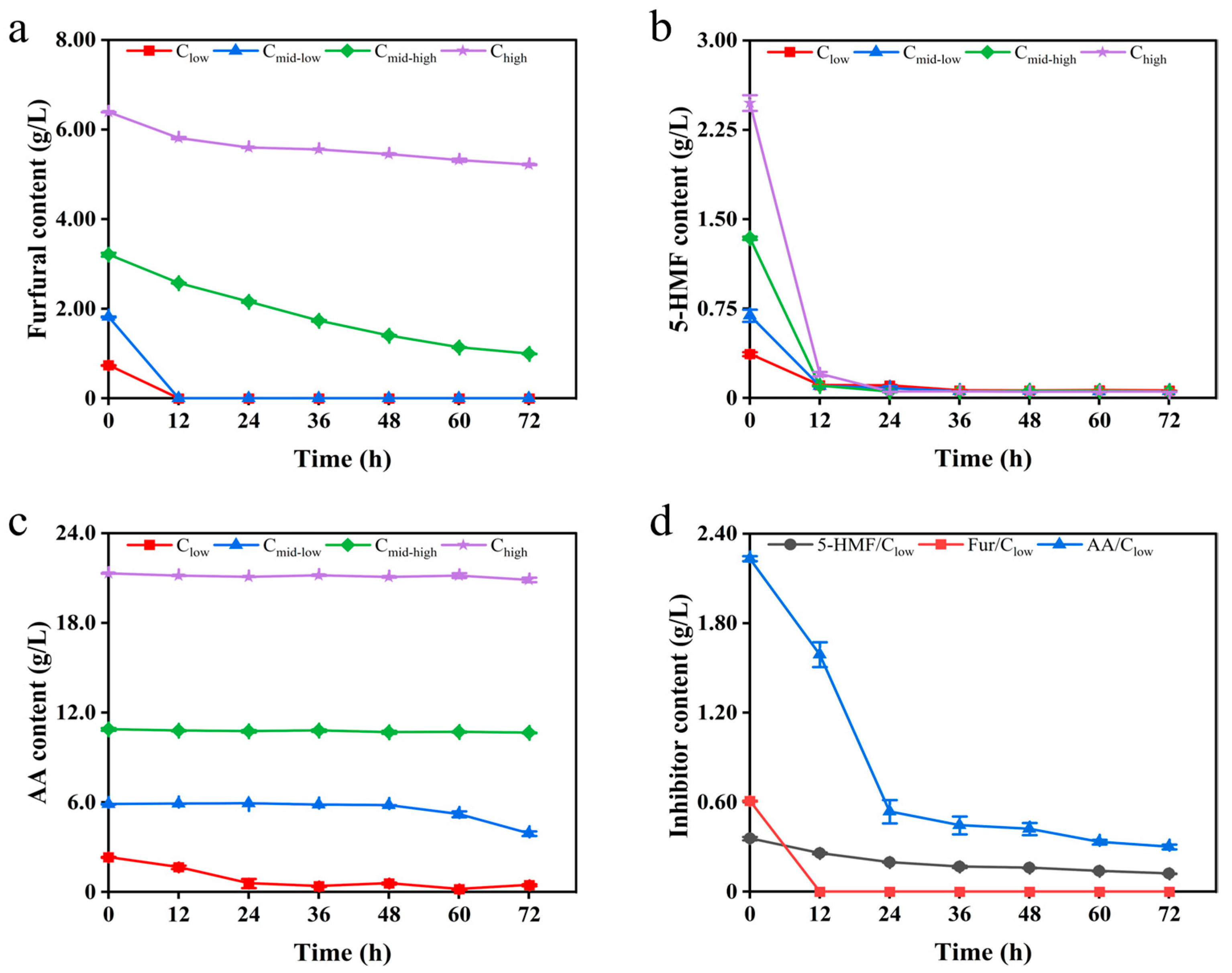
3.1. Growth Kinetics and Detoxification Characteristics of K. ohmeri SSK Under Stress of Inhibitors
3.2. Whole Genome Sequencing of K. ohmeri SSK
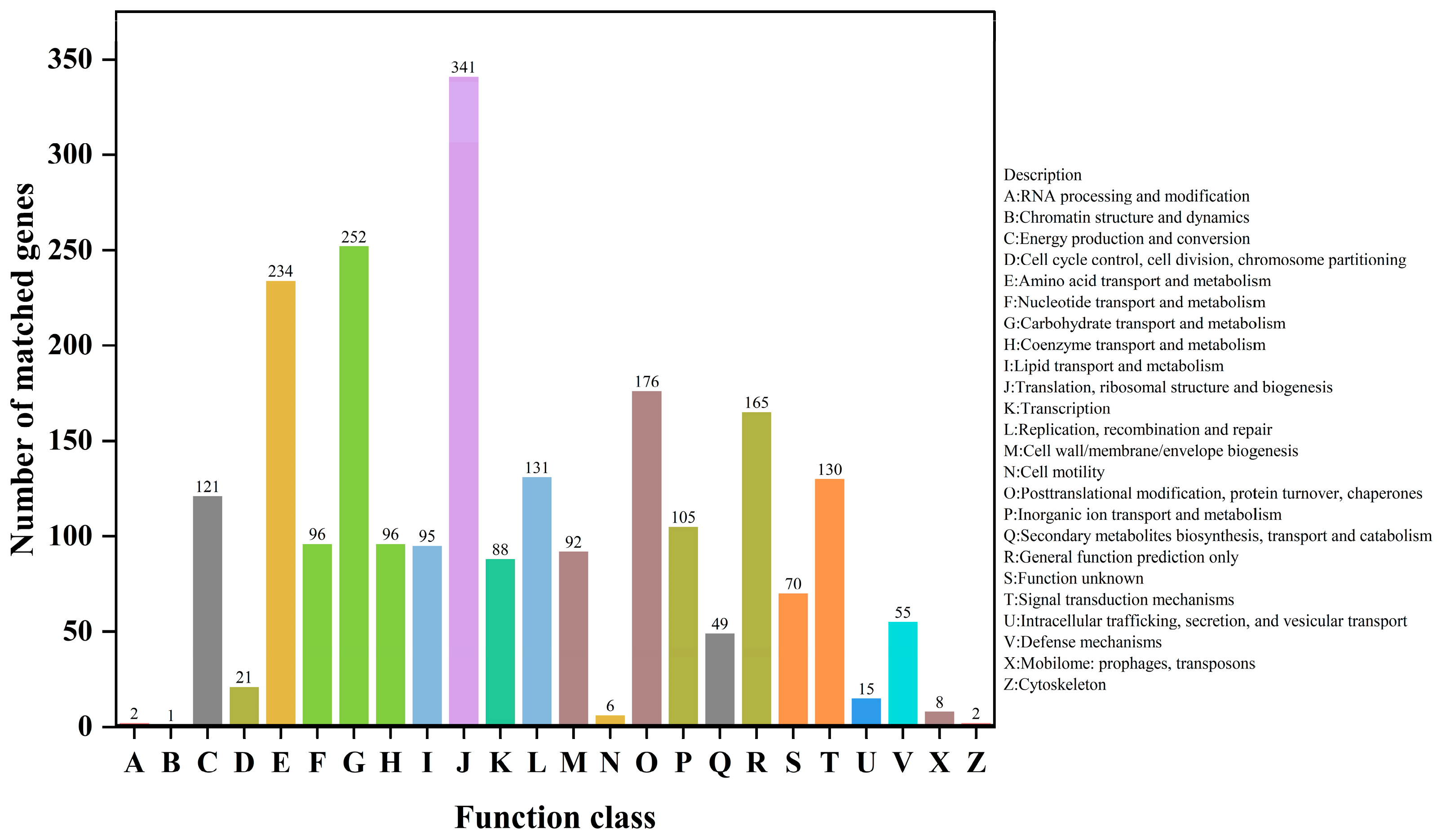
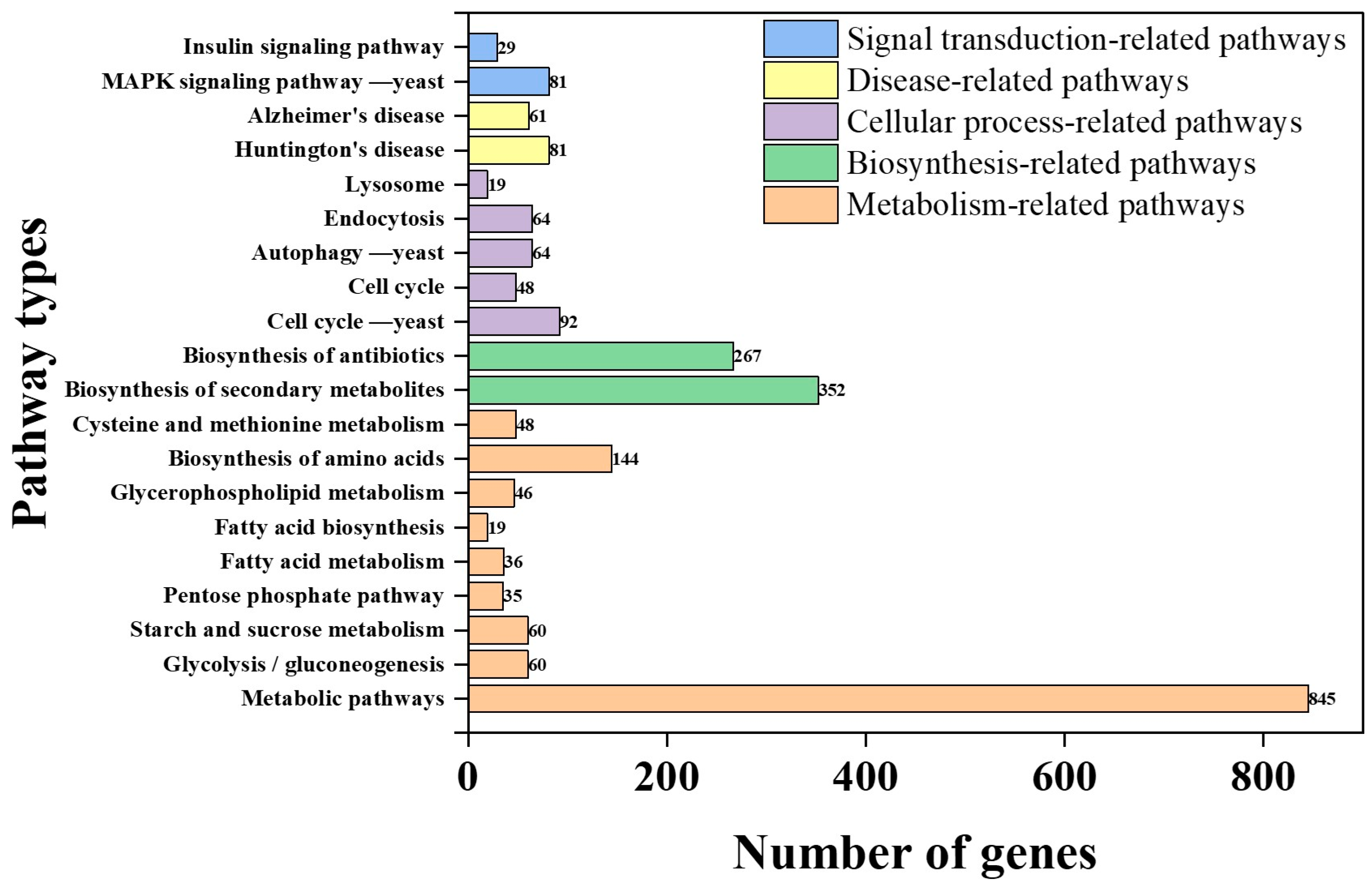
3.3. Gene Functional Annotation
3.4. Screening of Furfural and 5-HMF Degradation-Related Genes
3.5. ADH Sequence Analysis
3.6. Mining of Functional Genes of Acetic Acid Degradation Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, M.; Li, Y.; Kudinha, T.; Xu, Y.; Liu, Z. Kodamaea ohmeri as an emerging human pathogen: A review and update. Front. Microbiol. 2021, 12, 736582. [Google Scholar] [CrossRef]
- Liu, L.P.; Wang, Q.P.; Liu, J.; Quan, S.F.; Xie, D.C.; Hao, Y.E. Biological characteristics and virulence-related properties of a clinical Kodamaea ohmeri strain. J. Mycol. 2023, 42, 484–494. [Google Scholar] [CrossRef]
- Taj-Aldeen, S.J.; Doiphode, S.H.; Han, X.Y. Kodamaea (Pichia) ohmeri fungaemia in a premature neonate. J. Med. Microbiol. 2006, 55 Pt 2, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Liu, Z.Q.; Chi, Z.M. Production of phytase by a marine yeast Kodamaea ohmeri BG3 in an oats medium: Optimization by response surface methodology. Bioresour. Technol. 2008, 99, 6386–6390. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Y.; Xu, H.; Dai, X.Y.; Zhang, Y.; Ying, H.J.; Ouyang, P.K. Production of d-arabitol by a newly isolated Kodamaea ohmeri. Bioprocess. Biosyst. Eng. 2010, 33, 565–571. [Google Scholar] [CrossRef]
- Zhao, C.; Penttinen, P.; Zhang, L.; Dong, L.; Zhang, F.; Liao, D.; Zhang, S.; Li, Z.; Zhang, X. A novel high-level phenyllactic acid fungal producer, Kodamaea ohmeri w5 screened from fermented broad bean-chili-paste. Int. J. Food Microbiol. 2025, 426, 110923. [Google Scholar] [CrossRef]
- Heide-Marie, D.; Gino, V.; Takrama, J.F.; Nicholas, C.; Paul, D.V.; Luc, D.V. Yeast diversity of ghanaian cocoa bean heap fermentations. FEMS Yeast Res. 2010, 5, 774–783. [Google Scholar] [CrossRef]
- Cardoso, V.M.; Borelli, B.M.; Lara, C.A.; Soares, M.A.; Pataro, C.; Bodevan, E.C.; Rosa, C.A. The influence of seasons and ripening time on yeast communities of a traditional Brazilian cheese. Food Res. Int. 2015, 69, 331–340. [Google Scholar] [CrossRef]
- Xie, C.; Qin, L.; Liu, N.; Wen, A.; Zeng, H.; Miao, S. Flavor formation by amino acid catabolism in low-salt sufu paste, a Chinese fermented soybean food. Food Biosci. 2024, 59, 104228. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; He, F.; Pan, Z.; Du, Y.; Chen, Z.; He, Y.; Sun, Y.; Li, M. Effect of microbial communities on flavor profile of hakka rice wine throughout production. Food Chem. X 2021, 21, 101121. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, X.; Zhang, F.; Zhang, J. Impact of biodynamic and organic vineyard management on the microorganism community and aroma characteristics of cabernet sauvignon wine. J. Clean. Prod. 2024, 467, 142929. [Google Scholar] [CrossRef]
- Wang, F.; Ouyang, D.; Zhou, Z.; Page, S.J.; Liu, D.; Zhao, X. Lignocellulosic biomass as sustainable feedstock and materials for power generation and energy storage. J. Energy Chem. 2021, 57, 247–280. [Google Scholar] [CrossRef]
- Wierckx, N.; Koopman, F.; Ruijssenaars, H.J.; Winde, J.H. Microbial degradation of furanic compounds: Biochemistry, genetics, and impact. Appl. Microbiol. Biotechnol. 2011, 92, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Kłosowski, G.; Mikulski, D. Impact of lignocellulose pretreatment by-products on S. cerevisiae strain ethanol red metabolism during aerobic and an-aerobic growth. Molecules 2021, 26, 806. [Google Scholar] [CrossRef]
- Palmqvist, E.; Hahn-Hägerdal, B. Fermentation of lignocellulosic hydrolysates. II: Inhibitors and mechanisms of inhibition. Bioresour. Technol. 2000, 74, 25–33. [Google Scholar] [CrossRef]
- Sharma, S.; Arora, A.; Sharma, P.; Singh, S.; Nain, L.; Paul, D. Notable mixed substrate fermentation by native kodamaea ohmeri strains isolated from Lagenaria siceraria flowers and ethanol production on paddy straw hydrolysates. Chem. Cent. J. 2018, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Ujor, V.C.; Okonkwo, C.C. Microbial detoxification of lignocellulosic biomass hydrolysates: Biochemical and molecular aspects, challenges, exploits and future perspectives. Front. Bioeng. Biotechnol. 2022, 10, 1061667. [Google Scholar] [CrossRef]
- Trudgill, P.W. The metabolism of 2-furoic acid by Pseudomanas F2. Biochem. J. 1969, 113, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Koenig, K.; Andreesen, J.R. Xanthine dehydrogenase and 2-furoyl-coenzyme A dehydrogenase from Pseudomonas putida Fu1: Two molybdenum-containing dehydrogenases of novel structural composition. J. Bacteriol. 1990, 172, 5999–6009. [Google Scholar] [CrossRef] [PubMed]
- Koopman, F.; Wierckx, N.; Winde, J.H.; Ruijssenaars, H.J. Identification and characterization of the furfural and 5-(hydroxymethyl) furfural degradation pathways of Cupriavidus basilensis HMF14. Proc. Natl. Acad. Sci. USA 2010, 107, 4919–4924. [Google Scholar] [CrossRef]
- Koopman, F.; Wierckx, N.; Winde, J.H.; Ruijssenaars, H.J. Efficient whole-cell biotransformation of 5-(hydroxymethyl) furfural into FDCA, 2, 5-furandicarboxylic acid. Bioresour. Technol. 2010, 101, 6291–6296. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, Q.; Bao, J. Transcriptional analysis of Amorphotheca resinae ZN1 on biological degradation of furfural and 5-hydroxymethylfurfural derived from lignocellulose pretreatment. Biotechnol. Biofuels 2015, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Chew, K.L.; Achik, R.; Osman, N.H.; Octavia, S.; Teo, J.W.P. Genome sequence of a clinical blood isolate of Kodamaea ohmeri. Microbiol. Resour. Ann. 2022, 11, e00843-22. [Google Scholar] [CrossRef]
- Tauber, J.P.; Childers, A.K.; Evans, J.D. Draft genome sequence of the yeast Kodamaea ohmeri, a symbiont of the small hive beetle. Microbiol. Resour. Ann. 2019, 8, e00450-19. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.L.; Wang, X.Q.; Yuan, B.; Liu, C.G.; Zhao, X.Q. Manipulating cell flocculation-associated protein kinases in Saccharomyces cerevisiae enables improved stress tolerance and efficient cellulosic ethanol production. Bioresour. Technol. 2022, 348, 126758. [Google Scholar] [CrossRef]
- Zhang, F.; Ding, Y.; Zhu, C.D.; Zhou, X.; Orr, M.C.; Scheu, S.; Luan, Y.X. Phylogenomics from low-coverage whole-genome sequencing. Methods Ecol. Evol. 2019, 10, 507–517. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Guo, L.; Gu, S.; Wang, O.; Zhang, R.; Peters, B.A.; Fan, G.; Liu, X.; Xu, X.; Deng, L.; et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 2020, 9, giaa094. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, M.; Lomsadze, A. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr. Protoc. Bioinform. 2011, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Zahniser, M.P.; Prasad, S.; Kneen, M.M.; Kreinbring, C.A.; Petsko, G.A.; Ringe, D.; McLeish, M.J. Structure and mechanism of benzaldehyde dehydrogenase from Pseudomonas putida ATCC 12633, a member of the Class 3 aldehyde dehydrogenase superfamily. Protein Eng. Des. Sel. 2017, 30, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Calviño, E.; Carro, J.; Sánchez-Ruiz, M.I.; Cañada, F.J.; Martínez, A.T. Complete oxidation of hydroxymethylfurfural to furandicarboxylic acid by aryl-alcohol oxidase. Biotechnol. Biofuels 2019, 12, 217. [Google Scholar] [CrossRef]
- Wu, S.L.; Liu, Q.S.; Tan, H.D.; Zhang, F.Y.; Yi, H. Research progress on biocatalytic oxidation of 5-hydroxymethylfurfural. Biotechnol. Bull. 2016, 32, 50–58. [Google Scholar] [CrossRef]
- Shi, Z.G.; Ye, L.; Gong, P.T.; Zhao, D.G.; Liu, C.K. Analysis of alcohol dehydrogenase (ADH) family by bioinformatics. Genom. Appl. Biol. 2009, 28, 429–432. [Google Scholar] [CrossRef]
- Ausili, A.; Vitale, A.; Labella, T.; Rosso, F.; Barbarisi, A.; Gomez, J.C.; Auria, S. Alcohol dehydrogenase from the hyperthermophilic archaeon Pyrobaculum aerophilum: Stability at high temperature. Arch. Biochem. Biophys. 2012, 525, 40–46. [Google Scholar] [CrossRef]
- Shiba, Y.; Paradise, E.M.; Kirby, J.; Ro, D.K.; Keasling, J.D. Engineering of the pyruvate dehydrogenase bypass in Saccharomyces cerevisiae for high-level production of isoprenoids. Metab. Eng. 2007, 9, 160–168. [Google Scholar] [CrossRef]
- Deng, H.X.; Guo, C.C.; Li, E.H. Research progress on the metabolic regulation mechanism of acetic acid in Saccharomyces cerevisiae and the breeding of low—Yielding strains. Food Sci. 2023, 44, 183–192. [Google Scholar] [CrossRef]
- Yan, J.; Cheng, R.; Lin, X.; You, S.; Li, K.; Rong, H.; Ma, Y. Overexpression of acetyl-CoA synthetase increased the biomass and fatty acid proportion in microalga Schizochytrium. Appl. Microbiol. Biotechnol. 2013, 97, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Agyeman-Duah, E.; Ujor, V.C. Whole-genome sequence and fermentation characteristics of Enterobacter hormaechei UW0SKVC1: A promising candidate for detoxification of lignocellulosic biomass hydrolysates and production of value-added chemicals. Bioengineering 2023, 10, 1090. [Google Scholar] [CrossRef] [PubMed]
- Field, S.J.; Ryden, P.; Wilson, D.; James, S.A.; Roberts, I.N.; Richardson, D.J.; Waldron, K.W.; Clarke, T.A. Identification of furfural resistant strains of Saccharomyces cerevisiae and Saccharomyces paradoxus from a collection of environmental and industrial isolates. Biotechnol. Biofuels 2015, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.A.; Clark, W.; McCaffery, J.M.; Cai, Z.; Lanctot, A.; Slininger, P.J.; Liu, Z.L.; Gorsich, S.W. Furfural induces reactive oxygen species accumulation and cellular damage in Saccharomyces cerevisiae. Biotechnol. Biofuels 2010, 3, 2. [Google Scholar] [CrossRef]
- Gong, G.; Um, Y.; Park, T.H.; Woo, H.M. Complete genome sequence of Enterobacter cloacae GGT036: A furfural tolerant soil bacterium. J. Biotechnol. 2015, 193, 43–44. [Google Scholar] [CrossRef] [PubMed]
- Park, G.W.; Shin, S.; Kim, S.J.; Lee, J.S.; Moon, M.; Min, K. Rice straw-derived lipid production by HMF/furfural-tolerant oleaginous yeast generated by adaptive laboratory evolution. Bioresour. Technol. 2023, 367, 128220. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, M.; Masaki, K.; Fujii, T.; Iefuji, H. Yeast genes involved in response to lactic acid and acetic acid: Acidic conditions caused by the organic acids in Saccharomyces cerevisiae cultures induce expression of intracellular metal metabolism genes regulated by Aft1p. FEMS Yeast Res. 2006, 6, 924–936. [Google Scholar] [CrossRef]
- van Maris, A.J.; Abbott, D.A.; Bellissimi, E.; van den Brink, J.; Kuyper, M.; Luttik, M.A.; Wisselink, H.W.; Scheffers, W.A.; van Dijken, J.P.; Pronk, J.T. Alcoholic fermentation of carbon sources in biomass hydrolysates by Saccharomyces cerevisiae: Current status. Antonie Van. Leeuwenhoek. 2006, 90, 391–418. [Google Scholar] [CrossRef]
- Zhao, X.Q.; Zhang, M.M.; Xu, G.H.; Xu, J.R.; Bai, F.W. Progress in functional genomics of the molecular mechanism of acetic acid tolerance in Saccharomyces cerevisiae. Chin. J. Biotechnol. 2014, 30, 368–380. [Google Scholar] [CrossRef]
- Endo, A.; Tanizawa, Y.; Tanaka, N.; Maeno, S.; Kumar, H.; Shiwa, Y.; Okada, S.; Yoshikawa, H.; Dicks, L.; Nakagawa, J.; et al. Comparative genomics of Fructobacillus spp. and Leuconostoc spp. reveals niche-specific evolution of Fructobacillus spp. BMC Genom. 2015, 16, 1117. [Google Scholar] [CrossRef]
- Ma, W.; Yu, J.; Zhang, X.; Guo, S.; Zhang, F.; Jin, W.; Dong, J.; Jia, S.; Zhong, C.; Xue, J. Whole-genome sequencing exploitation analysis of non-Saccharomyces yeast Nakazawaea ishiwadae GDMCC 60786 and its physiological characterizations. Food Biosci. 2021, 41, 100982. [Google Scholar] [CrossRef]
- Demey, L.M.; Miller, C.R.; Manzella, M.P.; Spurbeck, R.R.; Sandhu, S.K.; Reguera, G.; Kashefi, K. The draft genome of the hyperthermophilic archaeon Pyrodictium delaneyi strain hulk, an iron and nitrate reducer, reveals the capacity for sulfate reduction. Stand. Genom. Sci. 2017, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.J.; Li, D.N.; Qiu, S.Y.; Zhou, J.Y.; Long, Z.H.; Wang, K.J.; Liu, M.Q.; Chen, J.; Cheng, D.; Pan, F.S. Identification and genome analysis of Laceyella sacchari FBKL4.014 isolated from moutai-flavor daqu. Food Sci. 2023, 44, 68–76. [Google Scholar] [CrossRef]
- Larroy, C.; Fernández, M.R.; González, E.; Parés, X.; Biosca, J.A. Characterization of the Saccharomyces cerevisiae YMR318C (ADH6) gene product as a broad specificity NADPH-dependent alcohol dehydrogenase: Relevance in aldehyde reduction. Biochem. J. 2002, 361 Pt 1, 163–172. [Google Scholar] [CrossRef]
- Ahmed-Laskar, A.; Younus, H. Aldehyde toxicity and metabolism: The role of aldehyde dehydrogenases in detoxification, drug resistance and carcinogenesis. Drug Metab. Rev. 2019, 51, 42–64. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metric | Value |
|---|---|
| Sample | K. ohmeri SSK |
| Total assembly length | 14,959,343 bp |
| GC content | 43.34% |
| Number of scaffolds | 457 |
| Scaffold N50 | 0.54 Mb |
| Scaffold L50 | 8 |
| N’s per 100 kbp | 4.29 |
| Strain | Genome Size (Mb) | GC (%) | N50 (Mb) | L50 | Number of Contigs | Source |
|---|---|---|---|---|---|---|
| K. ohmeri SSK | 14.3 | 43.34 | 0.5 | 8 | 457 | Bamboo |
| K. ohmeri 148 | 12.6 | 43.00 | 1.8 | 3 | 20 | Honeybee |
| K. ohmeri NRRL Y-1932 | 12.3 | 42.50 | 0.29 | 11 | 95 | NA |
| K. ohmeri W5 | 12.5 | 42.50 | 0.96 | 5 | 26 | Doubanjiang |
| K. ohmeri UWOPS05-228.2 | 12.3 | 42.50 | 0.76 | 6 | 89 | Bertam Palm |
| K. ohmeri 3873 | 12.3 | 43 | 0.36 | 11 | 104 | Homo sapiens |
| K. ohmeri UWOPS01-666b4 | 12.3 | 42.50 | 1.4 | 4 | 62 | Distimake tuberosus |
| K. ohmeri KO20 | 12.4 | 42.5 | 0.25 | 16 | 104 | Clinical Blood |
| K. ohmeri R6205-2 | 12.1 | 43.00 | 0.04 | 90 | 524 | Feces |
| Functional Categories | Annotation Description | Number |
|---|---|---|
| Alcohol dehydrogenase (31) | Zn-dependent alcohol dehydrogenase | 8 |
| NAD(P)+-dependent dehydrogenase, short-chain alcohol dehydrogenase | 16 | |
| Other alcohol dehydrogenase | 7 | |
| Aldehyde reductase, aldo/keto reductase (14) | Aldo/keto reductase | 12 |
| Aldehyde reductase | 2 | |
| aldehyde dehydrogenase (8) | NAD+-dependent aldehyde dehydrogenase | 6 |
| Semialdehyde dehydrogenase | 2 |
| Gene | Amino Acid Length | COG Best Hit Length | Alignment Length | Identity (%) | COG Best Hit Description | COG Class |
|---|---|---|---|---|---|---|
| scaffold3;1501_g | 500 | 428 | 241 | 29.0 | ACS family major facilitator superfamily protein | G |
| scaffold6;2737_g | 518 | 428 | 216 | 28.7 | ACS family major facilitator superfamily protein | G |
| scaffold6;2738_g | 582 | 491 | 201 | 25.9 | ACS family major facilitator superfamily protein | G |
| scaffold26;5535_g | 530 | 455 | 477 | 24.3 | ACS family major facilitator superfamily protein | G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.-Q.; Li, X.; Wang, Z.-F.; Deng, Y.-L.; Wang, Z.-Z.; Fang, X.-Y.; Zhang, M.-D.; Sun, W.; Zhao, X.-Q.; Liu, Z.-Q.; et al. Whole Genome Sequencing of Kodamaea ohmeri SSK and Its Characterization for Degradation of Inhibitors from Lignocellulosic Biomass. Biology 2025, 14, 458. https://doi.org/10.3390/biology14050458
Yang Y-Q, Li X, Wang Z-F, Deng Y-L, Wang Z-Z, Fang X-Y, Zhang M-D, Sun W, Zhao X-Q, Liu Z-Q, et al. Whole Genome Sequencing of Kodamaea ohmeri SSK and Its Characterization for Degradation of Inhibitors from Lignocellulosic Biomass. Biology. 2025; 14(5):458. https://doi.org/10.3390/biology14050458
Chicago/Turabian StyleYang, Yong-Qiang, Xu Li, Zhi-Fei Wang, Yu-Long Deng, Zhen-Zhi Wang, Xing-Yu Fang, Mao-Dong Zhang, Wei Sun, Xin-Qing Zhao, Zhi-Qiang Liu, and et al. 2025. "Whole Genome Sequencing of Kodamaea ohmeri SSK and Its Characterization for Degradation of Inhibitors from Lignocellulosic Biomass" Biology 14, no. 5: 458. https://doi.org/10.3390/biology14050458
APA StyleYang, Y.-Q., Li, X., Wang, Z.-F., Deng, Y.-L., Wang, Z.-Z., Fang, X.-Y., Zhang, M.-D., Sun, W., Zhao, X.-Q., Liu, Z.-Q., & Zhang, F.-L. (2025). Whole Genome Sequencing of Kodamaea ohmeri SSK and Its Characterization for Degradation of Inhibitors from Lignocellulosic Biomass. Biology, 14(5), 458. https://doi.org/10.3390/biology14050458