
Innate Lymphoid Cell Plasticity in Mucosal Infections

Abstract
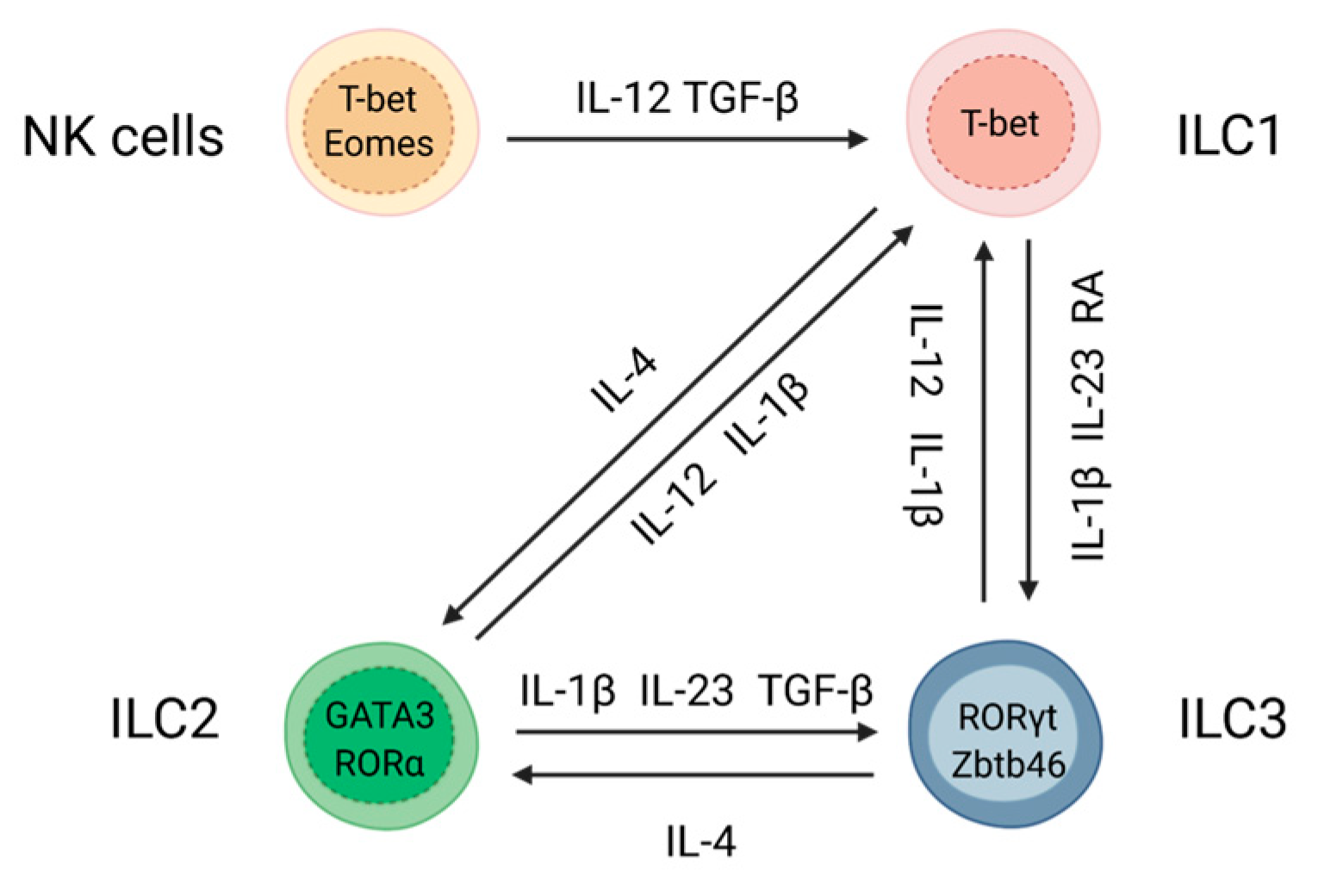
:1. Introduction
2. Role of Tissue Microenvironment in ILC Plasticity
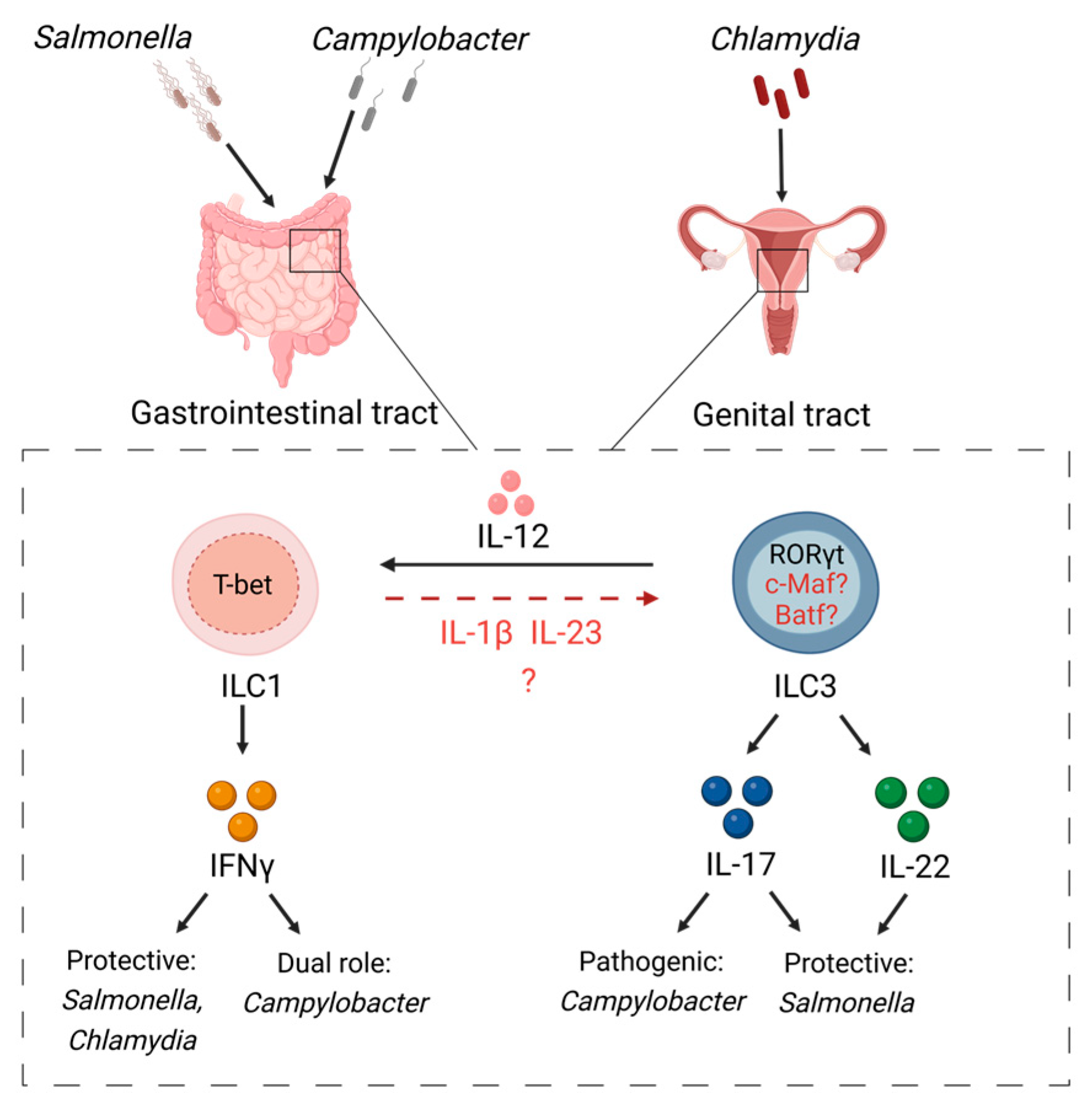
3. ILC Plasticity in Response to Gut Infections
4. Role of Gut Microbiota in ILC Plasticity
5. Role of Intestinal Hypoxia in ILC Plasticity
6. NK to ILC1 Plasticity in Response to T. gondii
7. ILC Plasticity in Response to Genital Tract Infections
8. ILC Plasticity in Response to Lung Infections
9. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Cherrier, M.; Ramachandran, G.; Golub, R. The interplay between innate lymphoid cells and T cells. Mucosal Immunol. 2020, 13, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Ngai, L.; Mortha, A.; Crome, S.Q. Tissue-Dependent Adaptations and Functions of Innate Lymphoid Cells. Front. Immunol. 2022, 13, 836999. [Google Scholar] [CrossRef] [PubMed]
- Korchagina, A.A.; Koroleva, E.; Tumanov, A.V. Innate Lymphoid Cells in Response to Intracellular Pathogens: Protection Versus Immunopathology. Front. Cell. Infect. Microbiol. 2021, 11, 775554. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Monticelli, L.A.; Fung, T.C.; Ziegler, C.G.; Grunberg, S.; Sinha, R.; Mantegazza, A.R.; Ma, H.L.; Crawford, A.; Angelosanto, J.M.; et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 2013, 498, 113–117. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science 2015, 348, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Magri, G.; Miyajima, M.; Bascones, S.; Mortha, A.; Puga, I.; Cassis, L.; Barra, C.M.; Comerma, L.; Chudnovskiy, A.; Gentile, M.; et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat. Immunol. 2014, 15, 354–364. [Google Scholar] [CrossRef]
- O’Connor, M.H.; Muir, R.; Chakhtoura, M.; Fang, M.; Moysi, E.; Moir, S.; Carey, A.J.; Terk, A.; Nichols, C.N.; Metcalf, T.; et al. A follicular regulatory Innate Lymphoid Cell population impairs interactions between germinal center Tfh and B cells. Commun. Biol. 2021, 4, 563. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Hepworth, M.R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 2019, 19, 599–613. [Google Scholar] [CrossRef]
- Shih, H.Y.; Sciumè, G.; Mikami, Y.; Guo, L.; Sun, H.W.; Brooks, S.R.; Urban, J.F., Jr.; Davis, F.P.; Kanno, Y.; O’Shea, J.J. Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 2016, 165, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.H.; Howard, J.K.; Lord, G.M. Transcription factor-driven regulation of ILC1 and ILC3. Trends Immunol. 2022, 43, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Fiancette, R.; Finlay, C.M.; Willis, C.; Bevington, S.L.; Soley, J.; Ng, S.T.H.; Baker, S.M.; Andrews, S.; Hepworth, M.R.; Withers, D.R. Reciprocal transcription factor networks govern tissue-resident ILC3 subset function and identity. Nat. Immunol. 2021, 22, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Herbert, D.R.; Douglas, B.; Zullo, K. Group 2 Innate Lymphoid Cells (ILC2): Type 2 Immunity and Helminth Immunity. Int. J. Mol. Sci. 2019, 20, 2276. [Google Scholar] [CrossRef]
- Seo, G.Y.; Giles, D.A.; Kronenberg, M. The role of innate lymphoid cells in response to microbes at mucosal surfaces. Mucosal Immunol. 2020, 13, 399–412. [Google Scholar] [CrossRef]
- Prager, I.; Liesche, C.; van Ooijen, H.; Urlaub, D.; Verron, Q.; Sandström, N.; Fasbender, F.; Claus, M.; Eils, R.; Beaudouin, J.; et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J. Exp. Med. 2019, 216, 2113–2127. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Artis, D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 2015, 21, 698–708. [Google Scholar] [CrossRef]
- Castro-Dopico, T.; Fleming, A.; Dennison, T.W.; Ferdinand, J.R.; Harcourt, K.; Stewart, B.J.; Cader, Z.; Tuong, Z.K.; Jing, C.; Lok, L.S.C.; et al. GM-CSF Calibrates Macrophage Defense and Wound Healing Programs during Intestinal Infection and Inflammation. Cell Rep. 2020, 32, 107857. [Google Scholar] [CrossRef]
- Pearson, C.; Thornton, E.E.; McKenzie, B.; Schaupp, A.L.; Huskens, N.; Griseri, T.; West, N.; Tung, S.; Seddon, B.P.; Uhlig, H.H.; et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. eLife 2016, 5, e10066. [Google Scholar] [CrossRef]
- Song, C.; Lee, J.S.; Gilfillan, S.; Robinette, M.L.; Newberry, R.D.; Stappenbeck, T.S.; Mack, M.; Cella, M.; Colonna, M. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J. Exp. Med. 2015, 212, 1869–1882. [Google Scholar] [CrossRef] [PubMed]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of innate lymphoid cell subsets. Nat. Rev. Immunol. 2020, 20, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Kabil, A.; Shin, S.B.; Hughes, M.R.; McNagny, K.M. “Just one word, plastic!”: Controversies and caveats in innate lymphoid cell plasticity. Front. Immunol. 2022, 13, 946905. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M. Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 2018, 48, 1104–1117. [Google Scholar] [CrossRef]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef]
- Lim, A.I.; Menegatti, S.; Bustamante, J.; Le Bourhis, L.; Allez, M.; Rogge, L.; Casanova, J.L.; Yssel, H.; Di Santo, J.P. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J. Exp. Med. 2016, 213, 569–583. [Google Scholar] [CrossRef]
- Park, E.; Patel, S.; Wang, Q.; Andhey, P.; Zaitsev, K.; Porter, S.; Hershey, M.; Bern, M.; Plougastel-Douglas, B.; Collins, P.; et al. Toxoplasma gondii infection drives conversion of NK cells into ILC1-like cells. eLife 2019, 8, e47605. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Ulland, T.K.; Cervantes-Barragan, L.; Bando, J.K.; Robinette, M.L.; Wang, Q.; White, A.J.; Gilfillan, S.; Cella, M.; Colonna, M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat. Immunol. 2017, 18, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Vonarbourg, C.; Mortha, A.; Bui, V.L.; Hernandez, P.P.; Kiss, E.A.; Hoyler, T.; Flach, M.; Bengsch, B.; Thimme, R.; Holscher, C.; et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 2010, 33, 736–751. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Otero, K.; Colonna, M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. USA 2010, 107, 10961–10966. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Khatun, A.; Kasmani, M.Y.; Chen, Y.; Zheng, S.; Atkinson, S.; Nguyen, C.; Burns, R.; Taparowsky, E.J.; Salzman, N.H.; et al. Group 3 innate lymphoid cells require BATF to regulate gut homeostasis in mice. J. Exp. Med. 2022, 219, e20211861. [Google Scholar] [CrossRef] [PubMed]
- Pokrovskii, M.; Hall, J.A.; Ochayon, D.E.; Yi, R.; Chaimowitz, N.S.; Seelamneni, H.; Carriero, N.; Watters, A.; Waggoner, S.N.; Littman, D.R.; et al. Characterization of Transcriptional Regulatory Networks that Promote and Restrict Identities and Functions of Intestinal Innate Lymphoid Cells. Immunity 2019, 51, 185–197.e6. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.E.; Barrera, A.; Wheaton, J.D.; Zuberbuehler, M.K.; Allan, D.S.J.; Carlyle, J.R.; Reddy, T.E.; Ciofani, M. c-Maf regulates the plasticity of group 3 innate lymphoid cells by restraining the type 1 program. J. Exp. Med. 2020, 217, e20191030. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, L.; Zhou, J.; Artis, D.; Longman, R.; Sonnenberg, G.F.; Scherl, E.; Sockolow, R.; Lukin, D.; Battat, R.; et al. ZBTB46 defines and regulates ILC3s that protect the intestine. Nature 2022, 609, 159–165. [Google Scholar] [CrossRef]
- Valle-Noguera, A.; Ochoa-Ramos, A.; Gomez-Sánchez, M.J.; Cruz-Adalia, A. Type 3 Innate Lymphoid Cells as Regulators of the Host-Pathogen Interaction. Front. Immunol. 2021, 12, 748851. [Google Scholar] [CrossRef] [PubMed]
- Bal, S.M.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; van Drunen, C.M.; Lutter, R.; Jonkers, R.E.; Hombrink, P.; et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 2016, 17, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Ohne, Y.; Silver, J.S.; Thompson-Snipes, L.; Collet, M.A.; Blanck, J.P.; Cantarel, B.L.; Copenhaver, A.M.; Humbles, A.A.; Liu, Y.J. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat. Immunol. 2016, 17, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Corral, D.; Charton, A.; Krauss, M.Z.; Blanquart, E.; Levillain, F.; Lefrançais, E.; Sneperger, T.; Vahlas, Z.; Girard, J.P.; Eberl, G.; et al. ILC precursors differentiate into metabolically distinct ILC1-like cells during Mycobacterium tuberculosis infection. Cell Rep. 2022, 39, 110715. [Google Scholar] [CrossRef]
- Golebski, K.; Ros, X.R.; Nagasawa, M.; van Tol, S.; Heesters, B.A.; Aglmous, H.; Kradolfer, C.M.A.; Shikhagaie, M.M.; Seys, S.; Hellings, P.W.; et al. IL-1β, IL-23, and TGF-β drive plasticity of human ILC2s towards IL-17-producing ILCs in nasal inflammation. Nat. Commun. 2019, 10, 2162. [Google Scholar] [CrossRef]
- Bernink, J.H.; Ohne, Y.; Teunissen, M.B.M.; Wang, J.; Wu, J.; Krabbendam, L.; Guntermann, C.; Volckmann, R.; Koster, J.; van Tol, S.; et al. c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nat. Immunol. 2019, 20, 992–1003. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Innate lymphoid cells in asthmatic patients. J. Allergy Clin. Immunol. 2019, 143, 1739–1741. [Google Scholar] [CrossRef]
- Silver, J.S.; Kearley, J.; Copenhaver, A.M.; Sanden, C.; Mori, M.; Yu, L.; Pritchard, G.H.; Berlin, A.A.; Hunter, C.A.; Bowler, R.; et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 2016, 17, 626–635. [Google Scholar] [CrossRef]
- Califano, D.; Furuya, Y.; Roberts, S.; Avram, D.; McKenzie, A.N.J.; Metzger, D.W. IFN-γ increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol. 2018, 11, 209–219. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, L.; Qiu, J.; Chen, X.; Hu-Li, J.; Siebenlist, U.; Williamson, P.R.; Urban, J.F., Jr.; Paul, W.E. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 2015, 16, 161–169. [Google Scholar] [CrossRef]
- Klose, C.S.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; d’Hargues, Y.; Goppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-bet gradient controls the fate and function of CCR6−RORgammat+ innate lymphoid cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef]
- Muraoka, W.T.; Korchagina, A.A.; Xia, Q.; Shein, S.A.; Jing, X.; Lai, Z.; Weldon, K.S.; Wang, L.J.; Chen, Y.; Kummer, L.W.; et al. Campylobacter infection promotes IFNgamma-dependent intestinal pathology via ILC3 to ILC1 conversion. Mucosal Immunol. 2021, 14, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Kirschnek, S.; Ortmann, N.; Tanriver, Y.; Hacker, G. The Reaction of Innate Lymphoid Cells in the Mouse Female Genital Tract to Chlamydial Infection. Infect. Immun. 2021, 89, e0080020. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, H.; Song, C.; Koprivsek, J.J.; Arulanandam, B.; Yang, H.; Tao, L.; Zhong, G. Adoptive Transfer of Group 3-Like Innate Lymphoid Cells Restores Mouse Colon Resistance to Colonization of a Gamma Interferon-Susceptible Chlamydia muridarum Mutant. Infect. Immun. 2021, 89, e00533-20. [Google Scholar] [CrossRef] [PubMed]
- Koprivsek, J.J.; He, Y.; Song, C.; Zhang, N.; Tumanov, A.; Zhong, G. Evasion of Innate Lymphoid Cell-Regulated Gamma Interferon Responses by Chlamydia muridarum To Achieve Long-Lasting Colonization in Mouse Colon. Infect. Immun. 2020, 88, e00798-19. [Google Scholar] [CrossRef]
- Hsu, R.J.; Yu, W.C.; Peng, G.R.; Ye, C.H.; Hu, S.; Chong, P.C.T.; Yap, K.Y.; Lee, J.Y.C.; Lin, W.C.; Yu, S.H. The Role of Cytokines and Chemokines in Severe Acute Respiratory Syndrome Coronavirus 2 Infections. Front. Immunol. 2022, 13, 832394. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168 e17. [Google Scholar] [CrossRef]
- Kim, Y.M.; Shin, E.C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021, 53, 750–760. [Google Scholar] [CrossRef]
- Silverstein, N.J.; Wang, Y.; Manickas-Hill, Z.; Carbone, C.; Dauphin, A.; Boribong, B.P.; Loiselle, M.; Davis, J.; Leonard, M.M.; Kuri-Cervantes, L.; et al. Innate lymphoid cells and COVID-19 severity in SARS-CoV-2 infection. eLife 2022, 11, e7468. [Google Scholar] [CrossRef]
- Lo Presti, E.; De Gaetano, A.; Pioggia, G.; Gangemi, S. Comprehensive Analysis of the ILCs and Unconventional T Cells in Virus Infection: Profiling and Dynamics Associated with COVID-19 Disease for a Future Monitoring System and Therapeutic Opportunities. Cells 2022, 11, 542. [Google Scholar] [CrossRef]
- Seo, G.Y.; Shui, J.W.; Takahashi, D.; Song, C.; Wang, Q.; Kim, K.; Mikulski, Z.; Chandra, S.; Giles, D.A.; Zahner, S.; et al. LIGHT-HVEM Signaling in Innate Lymphoid Cell Subsets Protects Against Enteric Bacterial Infection. Cell Host Microbe 2018, 24, 249–260.e4. [Google Scholar] [CrossRef] [PubMed]
- Besnard, A.G.; Guabiraba, R.; Niedbala, W.; Palomo, J.; Reverchon, F.; Shaw, T.N.; Couper, K.N.; Ryffel, B.; Liew, F.Y. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog. 2015, 11, e1004607. [Google Scholar] [CrossRef]
- Ng, S.S.; Souza-Fonseca-Guimaraes, F.; Rivera, F.L.; Amante, F.H.; Kumar, R.; Gao, Y.; Sheel, M.; Beattie, L.; Montes de Oca, M.; Guillerey, C.; et al. Rapid loss of group 1 innate lymphoid cells during blood stage Plasmodium infection. Clin. Transl. Immunol. 2018, 7, e1003. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Stevenson, M.M. Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect. Immun. 2000, 68, 4399–4406. [Google Scholar] [CrossRef] [PubMed]
- Abt, M.C.; Lewis, B.B.; Caballero, S.; Xiong, H.; Carter, R.A.; Sušac, B.; Ling, L.; Leiner, I.; Pamer, E.G. Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection against Acute Clostridium difficile Infection. Cell Host Microbe 2015, 18, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Krämer, B.; Goeser, F.; Lutz, P.; Glässner, A.; Boesecke, C.; Schwarze-Zander, C.; Kaczmarek, D.; Nischalke, H.D.; Branchi, V.; Manekeller, S.; et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017, 13, e1006373. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, L.; Zhao, J.; Li, G.; Zhang, L.; Chen, W.; Nie, W.; Reszka-Blanco, N.J.; Wang, F.S.; Su, L. Plasmacytoid dendritic cells promote HIV-1-induced group 3 innate lymphoid cell depletion. J. Clin. Investig. 2015, 125, 3692–3703. [Google Scholar] [CrossRef]
- Shah, S.V.; Manickam, C.; Ram, D.R.; Reeves, R.K. Innate Lymphoid Cells in HIV/SIV Infections. Front. Immunol. 2017, 8, 1818. [Google Scholar] [CrossRef]
- Kim, C.J.; Nazli, A.; Rojas, O.L.; Chege, D.; Alidina, Z.; Huibner, S.; Mujib, S.; Benko, E.; Kovacs, C.; Shin, L.Y.; et al. A role for mucosal IL-22 production and Th22 cells in HIV-associated mucosal immunopathogenesis. Mucosal Immunol. 2012, 5, 670–680. [Google Scholar] [CrossRef]
- Zeis, P.; Lian, M.; Fan, X.; Herman, J.S.; Hernandez, D.C.; Gentek, R.; Elias, S.; Symowski, C.; Knöpper, K.; Peltokangas, N.; et al. In Situ Maturation and Tissue Adaptation of Type 2 Innate Lymphoid Cell Progenitors. Immunity 2020, 53, 775–792.e9. [Google Scholar] [CrossRef]
- Ghaedi, M.; Shen, Z.Y.; Orangi, M.; Martinez-Gonzalez, I.; Wei, L.; Lu, X.; Das, A.; Heravi-Moussavi, A.; Marra, M.A.; Bhandoola, A.; et al. Single-cell analysis of RORα tracer mouse lung reveals ILC progenitors and effector ILC2 subsets. J. Exp. Med. 2020, 217, e20182293. [Google Scholar] [CrossRef] [PubMed]
- Chun, E.; Lavoie, S.; Fonseca-Pereira, D.; Bae, S.; Michaud, M.; Hoveyda, H.R.; Fraser, G.L.; Gallini Comeau, C.A.; Glickman, J.N.; Fuller, M.H.; et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity 2019, 51, 871–884.e6. [Google Scholar] [CrossRef] [PubMed]
- De Salvo, C.; Buela, K.A.; Pizarro, T.T. Cytokine-Mediated Regulation of Innate Lymphoid Cell Plasticity in Gut Mucosal Immunity. Front. Immunol. 2020, 11, 585319. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Sonnenberg, G.F. Activation and Suppression of Group 3 Innate Lymphoid Cells in the Gut. Trends Immunol. 2020, 41, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell 2016, 166, 1231–1246.e13. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Gamini, R.; Sécca, C.; Collins, P.L.; Zhao, S.; Peng, V.; Robinette, M.L.; Schettini, J.; Zaitsev, K.; Gordon, W.; et al. Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat. Immunol. 2019, 20, 980–991. [Google Scholar] [CrossRef]
- Nussbaum, K.; Burkhard, S.H.; Ohs, I.; Mair, F.; Klose, C.S.N.; Arnold, S.J.; Diefenbach, A.; Tugues, S.; Becher, B. Tissue microenvironment dictates the fate and tumor-suppressive function of type 3 ILCs. J. Exp. Med. 2017, 214, 2331–2347. [Google Scholar] [CrossRef]
- Yudanin, N.A.; Schmitz, F.; Flamar, A.L.; Thome, J.J.C.; Tait Wojno, E.; Moeller, J.B.; Schirmer, M.; Latorre, I.J.; Xavier, R.J.; Farber, D.L.; et al. Spatial and Temporal Mapping of Human Innate Lymphoid Cells Reveals Elements of Tissue Specificity. Immunity 2019, 50, 505–519.e4. [Google Scholar] [CrossRef]
- Robinette, M.L.; Fuchs, A.; Cortez, V.S.; Lee, J.S.; Wang, Y.; Durum, S.K.; Gilfillan, S.; Colonna, M. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol. 2015, 16, 306–317. [Google Scholar] [CrossRef]
- Kim, C.H.; Hashimoto-Hill, S.; Kim, M. Migration and Tissue Tropism of Innate Lymphoid Cells. Trends Immunol. 2016, 37, 68–79. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Tlaskalová-Hogenová, H.; Stěpánková, R.; Kozáková, H.; Hudcovic, T.; Vannucci, L.; Tučková, L.; Rossmann, P.; Hrnčíř, T.; Kverka, M.; Zákostelská, Z.; et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: Contribution of germ-free and gnotobiotic animal models of human diseases. Cell. Mol. Immunol. 2011, 8, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Denning, T.L.; Norris, B.A.; Medina-Contreras, O.; Manicassamy, S.; Geem, D.; Madan, R.; Karp, C.L.; Pulendran, B. Functional Specializations of Intestinal Dendritic Cell and Macrophage Subsets That Control Th17 and Regulatory T Cell Responses Are Dependent on the T Cell/APC Ratio, Source of Mouse Strain, and Regional Localization. J. Immunol. 2011, 187, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Satoh-Takayama, N.; Vosshenrich, C.A.J.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.-J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial Flora Drives Interleukin 22 Production in Intestinal NKp46+ Cells that Provide Innate Mucosal Immune Defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef]
- Chea, S.; Perchet, T.; Petit, M.; Verrier, T.; Guy-Grand, D.; Banchi, E.G.; Vosshenrich, C.A.; Di Santo, J.P.; Cumano, A.; Golub, R. Notch signaling in group 3 innate lymphoid cells modulates their plasticity. Sci. Signal. 2016, 9, ra45. [Google Scholar] [CrossRef]
- Viant, C.; Rankin, L.C.; Girard-Madoux, M.J.; Seillet, C.; Shi, W.; Smyth, M.J.; Bartholin, L.; Walzer, T.; Huntington, N.D.; Vivier, E.; et al. Transforming growth factor-β and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci. Signal. 2016, 9, ra46. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Golub, R. The Notch signaling pathway involvement in innate lymphoid cell biology. Biomed. J. 2021, 44, 133–143. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, X.; Pasha, M.A.; Siebel, C.W.; Costello, A.; Haczku, A.; MacNamara, K.; Liang, T.; Zhu, J.; Bhandoola, A.; et al. Cutting Edge: Notch Signaling Promotes the Plasticity of Group-2 Innate Lymphoid Cells. J. Immunol. 2017, 198, 1798–1803. [Google Scholar] [CrossRef]
- Li, S.; Bostick, J.W.; Ye, J.; Qiu, J.; Zhang, B.; Urban, J.F., Jr.; Avram, D.; Zhou, L. Aryl Hydrocarbon Receptor Signaling Cell Intrinsically Inhibits Intestinal Group 2 Innate Lymphoid Cell Function. Immunity 2018, 49, 915–928.e5. [Google Scholar] [CrossRef]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Kloverpris, H.N.; Kazer, S.W.; Mjosberg, J.; Mabuka, J.M.; Wellmann, A.; Ndhlovu, Z.; Yadon, M.C.; Nhamoyebonde, S.; Muenchhoff, M.; Simoni, Y.; et al. Innate Lymphoid Cells Are Depleted Irreversibly during Acute HIV-1 Infection in the Absence of Viral Suppression. Immunity 2016, 44, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lifshitz, L.; Gellatly, K.; Vinton, C.L.; Busman-Sahay, K.; McCauley, S.; Vangala, P.; Kim, K.; Derr, A.; Jaiswal, S.; et al. HIV-1-induced cytokines deplete homeostatic innate lymphoid cells and expand TCF7-dependent memory NK cells. Nat. Immunol. 2020, 21, 274–286. [Google Scholar] [CrossRef]
- Shalapour, S.; Deiser, K.; Sercan, O.; Tuckermann, J.; Minnich, K.; Willimsky, G.; Blankenstein, T.; Hämmerling, G.J.; Arnold, B.; Schüler, T. Commensal microflora and interferon-gamma promote steady-state interleukin-7 production in vivo. Eur. J. Immunol. 2010, 40, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, F.M.; von Burg, N.; Ivanek, R.; Teufel, C.; Horvath, E.; Peter, A.; Turchinovich, G.; Staehli, D.; Eichlisberger, T.; Gomez de Agüero, M.; et al. Microbiota-induced tissue signals regulate ILC3-mediated antigen presentation. Nat. Commun. 2020, 11, 1794. [Google Scholar] [CrossRef] [PubMed]
- Tizian, C.; Lahmann, A.; Hölsken, O.; Cosovanu, C.; Kofoed-Branzk, M.; Heinrich, F.; Mashreghi, M.F.; Kruglov, A.; Diefenbach, A.; Neumann, C. c-Maf restrains T-bet-driven programming of CCR6-negative group 3 innate lymphoid cells. eLife 2020, 9, e52549. [Google Scholar] [CrossRef] [PubMed]
- Krzywinska, E.; Sobecki, M.; Nagarajan, S.; Zacharjasz, J.; Tambuwala, M.M.; Pelletier, A.; Cummins, E.; Gotthardt, D.; Fandrey, J.; Kerdiles, Y.M.; et al. The transcription factor HIF-1α mediates plasticity of NKp46+ innate lymphoid cells in the gut. J. Exp. Med. 2022, 219, e20210909. [Google Scholar] [CrossRef] [PubMed]
- Hand, T.W.; Vujkovic-Cvijin, I.; Ridaura, V.K.; Belkaid, Y. Linking the Microbiota, Chronic Disease, and the Immune System. Trends Endocrinol. Metab. TEM 2016, 27, 831–843. [Google Scholar] [CrossRef]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.B.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.C.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013, 502, 96–99. [Google Scholar] [CrossRef]
- Sun, X.; Winglee, K.; Gharaibeh, R.Z.; Gauthier, J.; He, Z.; Tripathi, P.; Avram, D.; Bruner, S.; Fodor, A.; Jobin, C. Microbiota-Derived Metabolic Factors Reduce Campylobacteriosis in Mice. Gastroenterology 2018, 154, 1751–1763.e2. [Google Scholar] [CrossRef]
- Sun, X.; Threadgill, D.; Jobin, C. Campylobacter jejuni induces colitis through activation of mammalian target of rapamycin signaling. Gastroenterology 2012, 142, 86–95.e5. [Google Scholar] [CrossRef] [PubMed]
- Ganal-Vonarburg, S.C.; Duerr, C.U. The interaction of intestinal microbiota and innate lymphoid cells in health and disease throughout life. Immunology 2020, 159, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Sanos, S.L.; Bui, V.L.; Mortha, A.; Oberle, K.; Heners, C.; Johner, C.; Diefenbach, A. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 2009, 10, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Flach, M.; Mohle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef]
- Van Welden, S.; Selfridge, A.C.; Hindryckx, P. Intestinal hypoxia and hypoxia-induced signalling as therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 596–611. [Google Scholar] [CrossRef]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef]
- Karhausen, J.; Furuta, G.T.; Tomaszewski, J.E.; Johnson, R.S.; Colgan, S.P.; Haase, V.H. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Investig. 2004, 114, 1098–1106. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef]
- Robrahn, L.; Dupont, A.; Jumpertz, S.; Zhang, K.; Holland, C.H.; Guillaume, J.; Rappold, S.; Roth, J.; Cerovic, V.; Saez-Rodriguez, J.; et al. Stabilization but No Functional Influence of HIF-1α Expression in the Intestinal Epithelium during Salmonella Typhimurium Infection. Infect. Immun. 2022, 90, e00222-21. [Google Scholar] [CrossRef]
- Hirota, S.A.; Fines, K.; Ng, J.; Traboulsi, D.; Lee, J.; Ihara, E.; Li, Y.; Willmore, W.G.; Chung, D.; Scully, M.M.; et al. Hypoxia-inducible factor signaling provides protection in Clostridium difficile-induced intestinal injury. Gastroenterology 2010, 139, 259–269.e3. [Google Scholar] [CrossRef] [PubMed]
- Dowdell, A.S.; Cartwright, I.M.; Kitzenberg, D.A.; Kostelecky, R.E.; Mahjoob, O.; Saeedi, B.J.; Welch, N.; Glover, L.E.; Colgan, S.P. Essential role for epithelial HIF-mediated xenophagy in control of Salmonella infection and dissemination. Cell Rep. 2022, 40, 111409. [Google Scholar] [CrossRef]
- Di Luccia, B.; Gilfillan, S.; Cella, M.; Colonna, M.; Huang, S.C. ILC3s integrate glycolysis and mitochondrial production of reactive oxygen species to fulfill activation demands. J. Exp. Med. 2019, 216, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Fachi, J.L.; Pral, L.P.; Dos Santos, J.A.C.; Codo, A.C.; de Oliveira, S.; Felipe, J.S.; Zambom, F.F.F.; Câmara, N.O.S.; Vieira, P.; Colonna, M.; et al. Hypoxia enhances ILC3 responses through HIF-1α-dependent mechanism. Mucosal Immunol. 2021, 14, 828–841. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Teufel, C.; Horvath, E.; Peter, A.; Ercan, C.; Piscuoglio, S.; Hall, M.N.; Finke, D.; Lehmann, F.M. mTOR signaling mediates ILC3-driven immunopathology. Mucosal Immunol. 2021, 14, 1323–1334. [Google Scholar] [CrossRef]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef]
- Dunay, I.R.; Diefenbach, A. Group 1 innate lymphoid cells in Toxoplasma gondii infection. Parasite Immunol. 2018, 40, e12516. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Ehrentraut, S.; Bank, U.; Biswas, A.; Figueiredo, C.A.; Hölsken, O.; Düsedau, H.P.; Dovhan, V.; Knop, L.; Thode, J.; et al. Type 1 innate lymphoid cells regulate the onset of Toxoplasma gondii-induced neuroinflammation. Cell Rep. 2022, 38, 110564. [Google Scholar] [CrossRef] [PubMed]
- Dunay, I.R.; Damatta, R.A.; Fux, B.; Presti, R.; Greco, S.; Colonna, M.; Sibley, L.D. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 2008, 29, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Bajénoff, M.; Breart, B.; Huang, A.Y.; Qi, H.; Cazareth, J.; Braud, V.M.; Germain, R.N.; Glaichenhaus, N. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J. Exp. Med. 2006, 203, 619–631. [Google Scholar] [CrossRef]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef]
- Willinger, T. Metabolic Control of Innate Lymphoid Cell Migration. Front. Immunol. 2019, 10, 2010. [Google Scholar] [CrossRef]
- Dutton, E.E.; Gajdasik, D.W.; Willis, C.; Fiancette, R.; Bishop, E.L.; Camelo, A.; Sleeman, M.A.; Coccia, M.; Didierlaurent, A.M.; Tomura, M.; et al. Peripheral lymph nodes contain migratory and resident innate lymphoid cell populations. Sci. Immunol. 2019, 4, eaau8082. [Google Scholar] [CrossRef]
- Zhang, J.; Qiu, J.; Zhou, W.; Cao, J.; Hu, X.; Mi, W.; Su, B.; He, B.; Qiu, J.; Shen, L. Neuropilin-1 mediates lung tissue-specific control of ILC2 function in type 2 immunity. Nat. Immunol. 2022, 23, 237–250. [Google Scholar] [CrossRef]
- Shikhagaie, M.M.; Björklund, Å.K.; Mjösberg, J.; Erjefält, J.S.; Cornelissen, A.S.; Ros, X.R.; Bal, S.M.; Koning, J.J.; Mebius, R.E.; Mori, M.; et al. Neuropilin-1 Is Expressed on Lymphoid Tissue Residing LTi-like Group 3 Innate Lymphoid Cells and Associated with Ectopic Lymphoid Aggregates. Cell Rep. 2017, 18, 1761–1773. [Google Scholar] [CrossRef]
- Goldszmid, R.S.; Caspar, P.; Rivollier, A.; White, S.; Dzutsev, A.; Hieny, S.; Kelsall, B.; Trinchieri, G.; Sher, A. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 2012, 36, 1047–1059. [Google Scholar] [CrossRef]
- Lopez-Yglesias, A.H.; Burger, E.; Camanzo, E.; Martin, A.T.; Araujo, A.M.; Kwok, S.F.; Yarovinsky, F. T-bet-dependent ILC1- and NK cell-derived IFN-gamma mediates cDC1-dependent host resistance against Toxoplasma gondii. PLoS Pathog. 2021, 17, e1008299. [Google Scholar] [CrossRef] [PubMed]
- Goldszmid, R.S.; Bafica, A.; Jankovic, D.; Feng, C.G.; Caspar, P.; Winkler-Pickett, R.; Trinchieri, G.; Sher, A. TAP-1 indirectly regulates CD4+ T cell priming in Toxoplasma gondii infection by controlling NK cell IFN-gamma production. J. Exp. Med. 2007, 204, 2591–2602. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Arranz-Solís, D.; Saeij, J.P.J. Influence of the Host and Parasite Strain on the Immune Response During Toxoplasma Infection. Front. Cell. Infect. Microbiol. 2020, 10, 580425. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, D.L.; Krempels, R.; Denton, S.L.; Fettel, K.D.; Saltz, G.M.; Rach, D.; Fatima, R.; Mundhenke, T.; Materi, J.; Dunay, I.R.; et al. NK Cells Negatively Regulate CD8 T Cells to Promote Immune Exhaustion and Chronic Toxoplasma gondii Infection. Front. Cell. Infect. Microbiol. 2020, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Monin, L.; Whettlock, E.M.; Male, V. Immune responses in the human female reproductive tract. Immunology 2020, 160, 106–115. [Google Scholar] [CrossRef]
- Doisne, J.M.; Balmas, E.; Boulenouar, S.; Gaynor, L.M.; Kieckbusch, J.; Gardner, L.; Hawkes, D.A.; Barbara, C.F.; Sharkey, A.M.; Brady, H.J.; et al. Composition, Development, and Function of Uterine Innate Lymphoid Cells. J. Immunol. 2015, 195, 3937–3945. [Google Scholar] [CrossRef] [PubMed]
- Vacca, P.; Montaldo, E.; Croxatto, D.; Loiacono, F.; Canegallo, F.; Venturini, P.L.; Moretta, L.; Mingari, M.C. Identification of diverse innate lymphoid cells in human decidua. Mucosal Immunol. 2015, 8, 254–264. [Google Scholar] [CrossRef]
- Xu, H.; Su, X.; Zhao, Y.; Tang, L.; Chen, J.; Zhong, G. Innate Lymphoid Cells Are Required for Endometrial Resistance to Chlamydia trachomatis Infection. Infect. Immun. 2020, 88, e00152-20. [Google Scholar] [CrossRef]
- Mercado, M.A.B.; Du, W.; Malaviarachchi, P.A.; Gann, J.I.; Li, L.X. Innate IFN-gamma Is Essential for Systemic Chlamydia muridarum Control in Mice, While CD4 T Cell-Dependent IFN-gamma Production Is Highly Redundant in the Female Reproductive Tract. Infect. Immun. 2021, 89, e00541-20. [Google Scholar] [CrossRef]
- Raimondi, S.; Candeliere, F.; Amaretti, A.; Foschi, C.; Morselli, S.; Gaspari, V.; Rossi, M.; Marangoni, A. Vaginal and Anal Microbiome during Chlamydia trachomatis Infections. Pathogens 2021, 10, 1347. [Google Scholar] [CrossRef]
- Ziklo, N.; Vidgen, M.E.; Taing, K.; Huston, W.M.; Timms, P. Dysbiosis of the Vaginal Microbiota and Higher Vaginal Kynurenine/Tryptophan Ratio Reveals an Association with Chlamydia trachomatis Genital Infections. Front. Cell. Infect. Microbiol. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Helble, J.D.; Reinhold-Larsson, N.V.; Starnbach, M.N. Early Colonization of the Upper Genital Tract by Chlamydia muridarum Is Associated with Enhanced Inflammation Later in Infection. Infect. Immun. 2019, 87, e00405-19. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Song, X.; Wei, W.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 8, 875. [Google Scholar] [CrossRef] [PubMed]
- De Grove, K.C.; Provoost, S.; Verhamme, F.M.; Bracke, K.R.; Joos, G.F.; Maes, T.; Brusselle, G.G. Characterization and Quantification of Innate Lymphoid Cell Subsets in Human Lung. PLoS ONE 2016, 11, e0145961. [Google Scholar] [CrossRef]
- Puttur, F.; Denney, L.; Gregory, L.G.; Vuononvirta, J.; Oliver, R.; Entwistle, L.J.; Walker, S.A.; Headley, M.B.; McGhee, E.J.; Pease, J.E.; et al. Pulmonary environmental cues drive group 2 innate lymphoid cell dynamics in mice and humans. Sci. Immunol. 2019, 4, eaav7638. [Google Scholar] [CrossRef]
- Miller, M.M.; Patel, P.S.; Bao, K.; Danhorn, T.; O’Connor, B.P.; Reinhardt, R.L. BATF acts as an essential regulator of IL-25-responsive migratory ILC2 cell fate and function. Sci. Immunol. 2020, 5, eaay3994. [Google Scholar] [CrossRef]
- Lim, A.I.; Li, Y.; Lopez-Lastra, S.; Stadhouders, R.; Paul, F.; Casrouge, A.; Serafini, N.; Puel, A.; Bustamante, J.; Surace, L.; et al. Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation. Cell 2017, 168, 1086–1100.e10. [Google Scholar] [CrossRef]
- Croft, C.A.; Thaller, A.; Marie, S.; Doisne, J.-M.; Surace, L.; Yang, R.; Puel, A.; Bustamante, J.; Casanova, J.-L.; Di Santo, J.P. Notch, RORC and IL-23 signals cooperate to promote multi-lineage human innate lymphoid cell differentiation. Nat. Commun. 2022, 13, 4344. [Google Scholar] [CrossRef]
- Lim, A.I.; Di Santo, J.P. ILC-poiesis: Ensuring tissue ILC differentiation at the right place and time. Eur. J. Immunol. 2019, 49, 11–18. [Google Scholar] [CrossRef]
- Harly, C.; Kenney, D.; Ren, G.; Lai, B.; Raabe, T.; Yang, Q.; Cam, M.C.; Xue, H.H.; Zhao, K.; Bhandoola, A. The transcription factor TCF-1 enforces commitment to the innate lymphoid cell lineage. Nat. Immunol. 2019, 20, 1150–1160. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, M.; Dai, M.; Yue, S.; Li, Z.; Qiu, J.; Lu, C.; Xu, W. IL-18/IL-18R Signaling Is Dispensable for ILC Development But Constrains the Growth of ILCP/ILCs. Front. Immunol. 2022, 13, 923424. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.E.; Smith, J.N.P.; Fredman, G.; MacNamara, K.C. IL-18R-mediated HSC quiescence and MLKL-dependent cell death limit hematopoiesis during infection-induced shock. Stem Cell Rep. 2021, 16, 2887–2899. [Google Scholar] [CrossRef] [PubMed]
- Ihim, S.A.; Abubakar, S.D.; Zian, Z.; Sasaki, T.; Saffarioun, M.; Maleknia, S.; Azizi, G. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity: Biological role in induction, regulation, and treatment. Front. Immunol. 2022, 13, 919973. [Google Scholar] [CrossRef]
- Ardain, A.; Domingo-Gonzalez, R.; Das, S.; Kazer, S.W.; Howard, N.C.; Singh, A.; Ahmed, M.; Nhamoyebonde, S.; Rangel-Moreno, J.; Ogongo, P.; et al. Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature 2019, 570, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Kim, M.H.; Friesen, L.; Kim, C.H. BATF regulates innate lymphoid cell hematopoiesis and homeostasis. Sci. Immunol. 2020, 5, eaaz8154. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kasmani, M.Y.; Zheng, S.; Khatun, A.; Chen, Y.; Winkler, W.; Zander, R.; Burns, R.; Taparowsky, E.J.; Sun, J.; et al. BATF promotes group 2 innate lymphoid cell&-mediated lung tissue protection during acute respiratory virus infection. Sci. Immunol. 2022, 7, eabc9934. [Google Scholar] [PubMed]
- Shen, W.; Huang, J.; Wang, Y. Biological Significance of NOTCH Signaling Strength. Front. Cell Dev. Biol. 2021, 9, 652273. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Cao, W.; Endrias, K.; Kuchipudi, S.V.; Mittal, S.K.; Sambhara, S. Innate lymphoid cells (ILC) in SARS-CoV-2 infection. Mol. Asp. Med. 2021, 80, 101008. [Google Scholar] [CrossRef]
- Gomez-Cadena, A.; Spehner, L.; Kroemer, M.; Khelil, M.B.; Bouiller, K.; Verdeil, G.; Trabanelli, S.; Borg, C.; Loyon, R.; Jandus, C. Severe COVID-19 patients exhibit an ILC2 NKG2D(+) population in their impaired ILC compartment. Cell. Mol. Immunol. 2021, 18, 484–486. [Google Scholar] [CrossRef]
- Laffont, S.; Blanquart, E.; Savignac, M.; Cénac, C.; Laverny, G.; Metzger, D.; Girard, J.P.; Belz, G.T.; Pelletier, L.; Seillet, C.; et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1581–1592. [Google Scholar] [CrossRef]
- Cephus, J.Y.; Stier, M.T.; Fuseini, H.; Yung, J.A.; Toki, S.; Bloodworth, M.H.; Zhou, W.; Goleniewska, K.; Zhang, J.; Garon, S.L.; et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep. 2017, 21, 2487–2499. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ILC Plasticity | Tissue | Pathogen | Type of Pathogen | Main Cytokine and Function |
|---|---|---|---|---|
| NK→ILC1 | Spleen, lungs, liver | Toxoplasma gondii | Protozoan parasite | Protective IFNγ [32] |
| ILC2→ILC1 | Lungs | Mycobacterium tuberculosis | Intracellular bacteria | Protective IFNγ [44] |
| Lungs | Influenza virus | Respiratory virus | Protective/pathogenic IFNγ [48,49] | |
| ILC2→ILC3 | Skin, Tongue | Candida albicans | Fungus | Protective IL-17 [46,50] |
| Intestine, lungs | Nippostrongylus brasiliensis | Helminth | Protective/pathogenic IL-17 [50] | |
| ILC3→ILC1 | Intestine | Salmonella typhimurium | Intracellular bacteria | Protective/pathogenic IFNγ [51] |
| Intestine | Campylobacter jejuni | Intracellular bacteria | Pathogenic/protective IFNγ [52] | |
| Genital tract | Chlamydia spp. | Intracellular bacteria | Protective IFNγ [53,54,55] | |
| ILC plasticity not yet determined | Lungs | SARS-CoV-2 | Respiratory virus | Pathogenic/protective IFN type I-III, TNF, IL-1b, CCL2 [56,57,58,59,60] |
| Intestine | Yersinia enterocolitica | Intracellular bacteria | Protective IFNγ [61] | |
| Spleen, liver | Plasmodium spp. | Protozoan parasite | Protective IL-4/IL-5/IL-13 [62]; Protective IFNγ [63,64] | |
| Intestine | Clostridium difficile | Extracellular bacteria | Protective IFNγ/IL-22 [65] | |
| Intestine | HIV-1 | Virus | Protective IL-22 [66,67,68,69] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korchagina, A.A.; Koroleva, E.; Tumanov, A.V. Innate Lymphoid Cell Plasticity in Mucosal Infections. Microorganisms 2023, 11, 461. https://doi.org/10.3390/microorganisms11020461
Korchagina AA, Koroleva E, Tumanov AV. Innate Lymphoid Cell Plasticity in Mucosal Infections. Microorganisms. 2023; 11(2):461. https://doi.org/10.3390/microorganisms11020461
Chicago/Turabian StyleKorchagina, Anna A., Ekaterina Koroleva, and Alexei V. Tumanov. 2023. "Innate Lymphoid Cell Plasticity in Mucosal Infections" Microorganisms 11, no. 2: 461. https://doi.org/10.3390/microorganisms11020461