Identifying Protein–metabolite Networks Associated with COPD Phenotypes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Introduction
2.2. Correlations between Adjusted -Omic Data and Phenotype
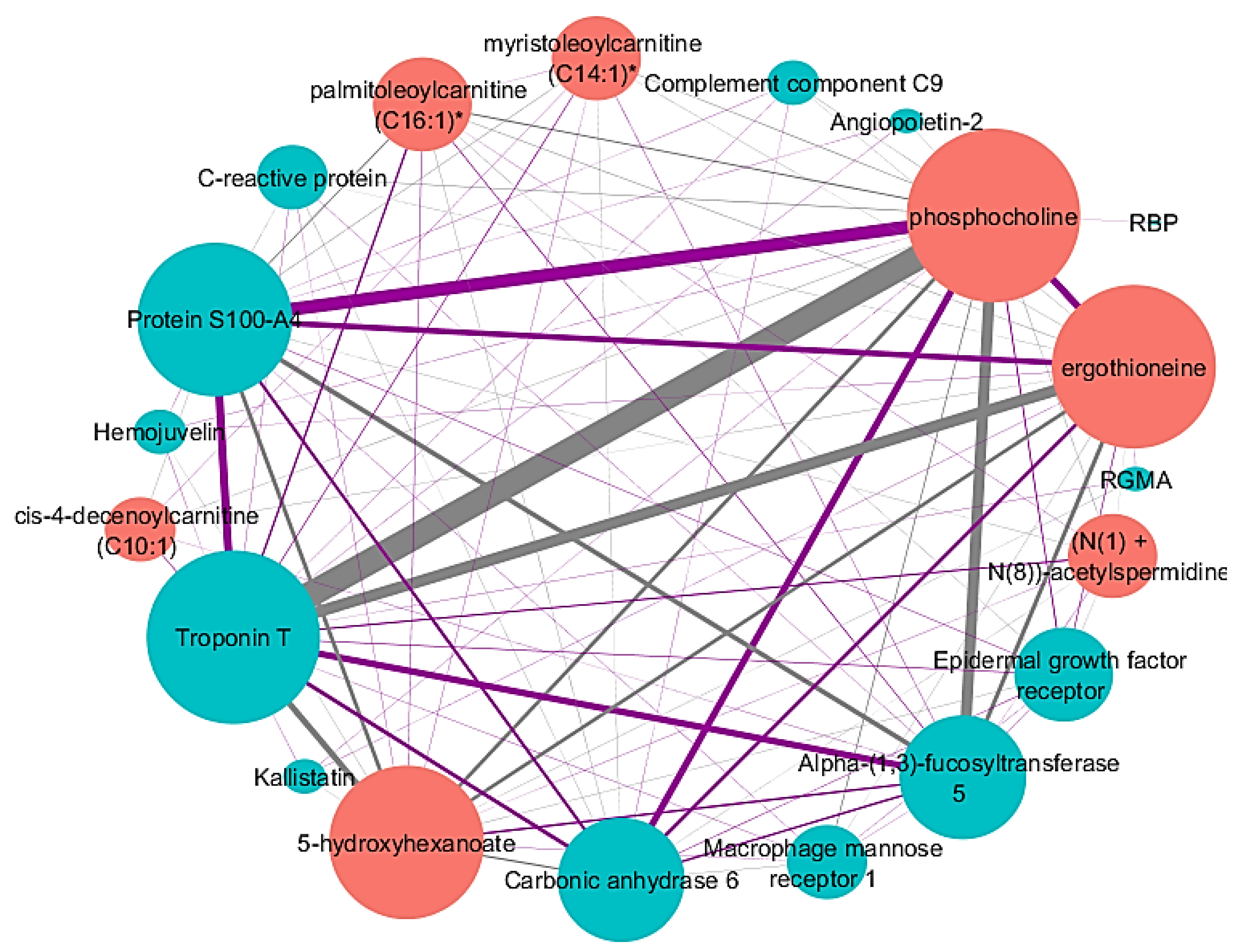
2.3. Identified Network Associated with FEV1%
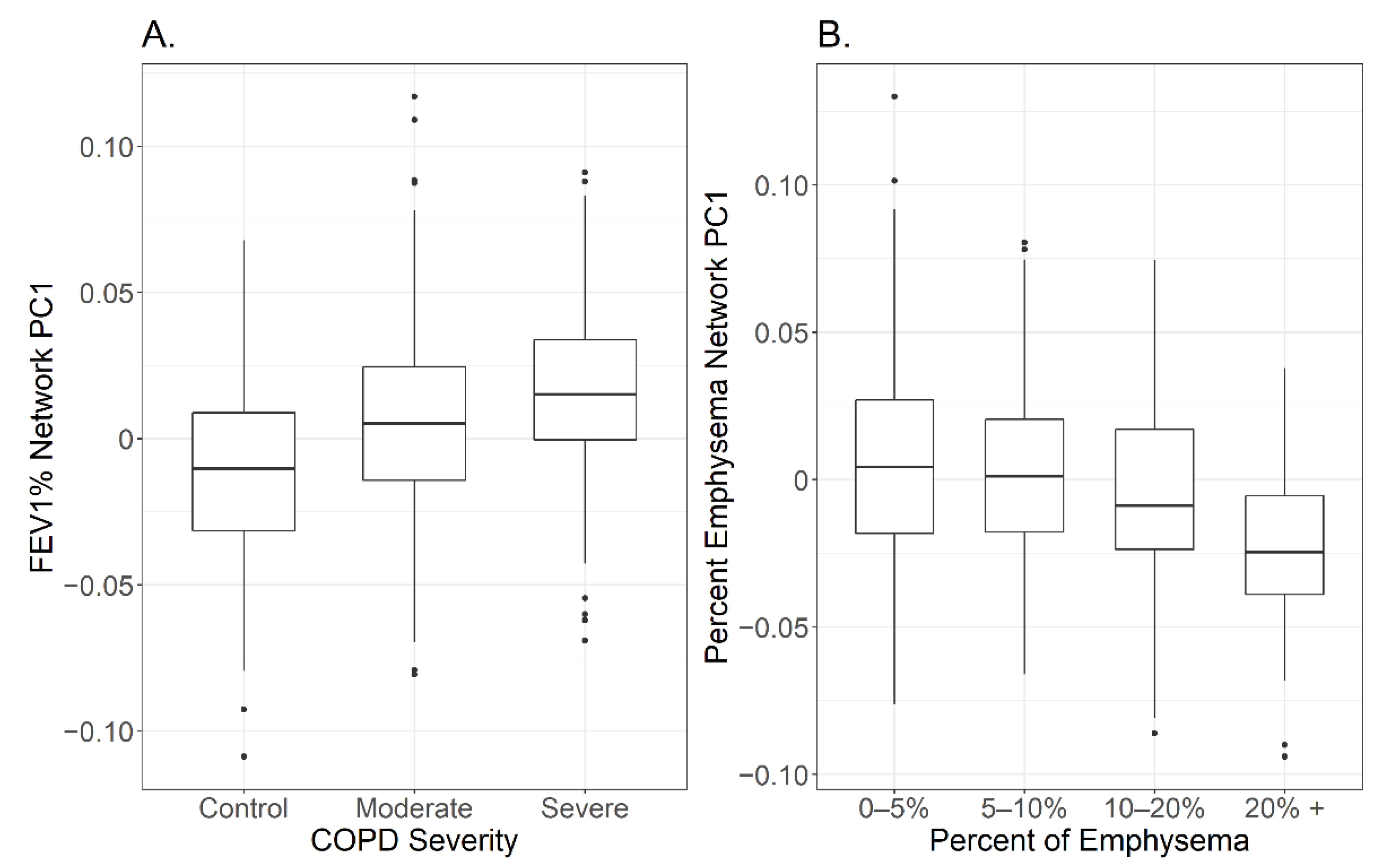
2.4. Network Comparison between Adjusted and Unadjusted -Omic Data with FEV1%
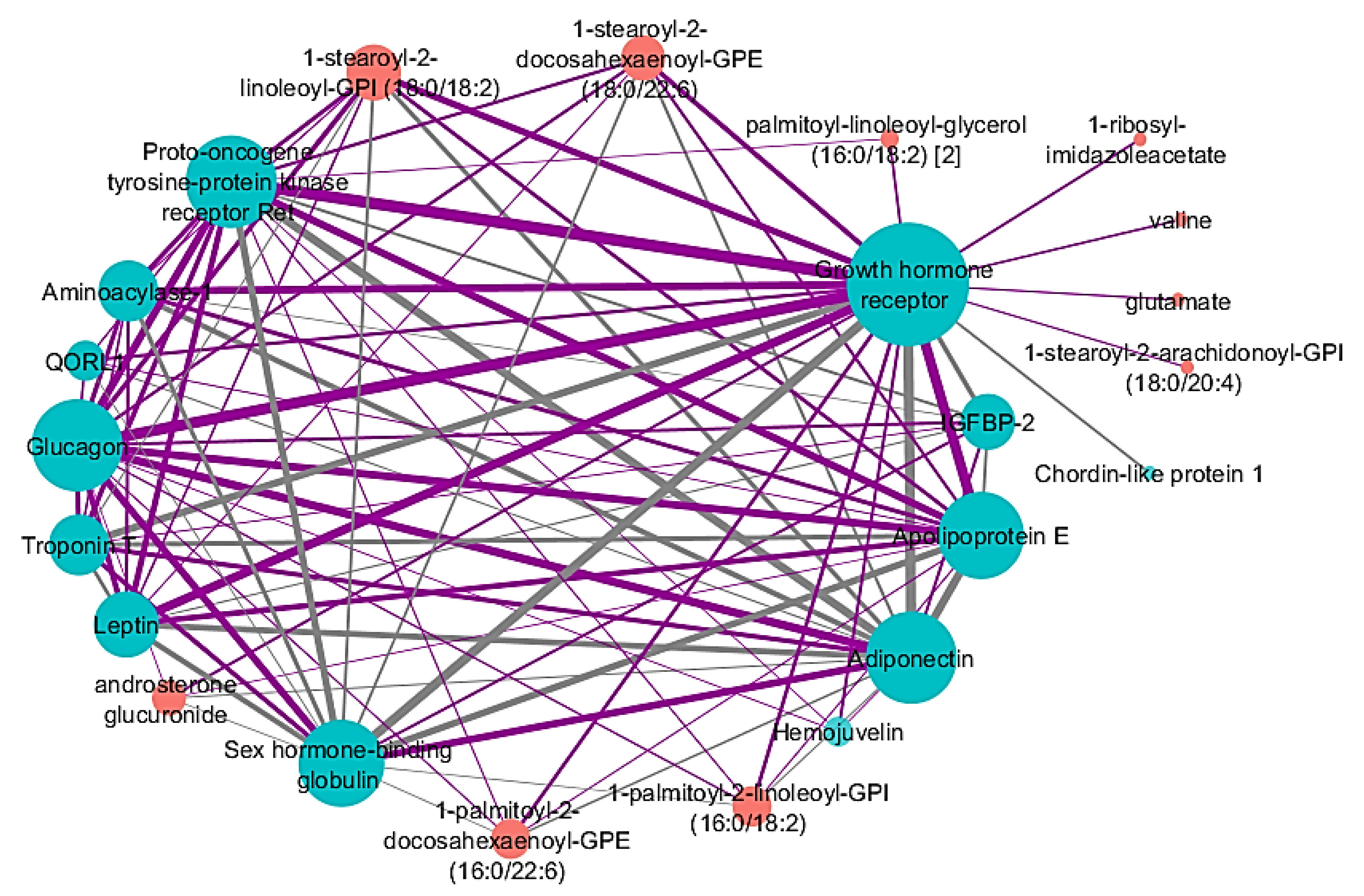
2.5. Identified Network Associated with Percent Emphysema
2.6. Network Comparison between Adjusted and Unadjusted -Omic Data with Percent Emphysema
2.7. Secondary Network Analysis
3. Discussion
4. Materials and Methods
4.1. COPDGene
4.2. Clinical Variables and Definitions
4.3. Proteomics and Data Processing
4.4. Metabolomics and Data Processing
4.5. Adjusted Proteomic and Metabolomic Data
4.6. Statistical Package
4.7. SmCCNet
4.8. Manual Hyperparameter Optimization Process
4.9. Final Protein–Metabolite Network Correlations
4.10. Network Sensitivity Analysis
4.11. Secondary Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garcia, I.F.F.; Tiuganji, T.G.; Morais Pereira Simões, M.d.S.; Santoro, I.L.; Lunardi, A.C. Systemic effects of chronic obstructive pulmonary disease in young-old adults’ life-space mobility. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 2777–2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzikhan, N.; Verhamme, K.M.C.; Hofman, A.; Stricker, B.H.; Brusselle, G.G.; Lahousse, L. Prevalence and incidence of COPD in smokers and non-smokers: the Rotterdam Study. Eur. J. Epidemiol. 2016, 31, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedlander, A.L.; Lynch, D.; Dyar, L.A.; Bowler, R.P. Phenotypes of chronic obstructive pulmonary disease. COPD 2007, 4, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Carolan, B.J.; Hughes, G.; Morrow, J.; Hersh, C.P.; O’Neal, W.K.; Rennard, S.; Pillai, S.G.; Belloni, P.; Cockayne, D.A.; Comellas, A.P.; et al. The association of plasma biomarkers with computed tomography-assessed emphysema phenotypes. Respir. Res. 2014, 15, 127. [Google Scholar] [CrossRef] [Green Version]
- Takei, N.; Suzuki, M.; Makita, H.; Konno, H.; Shimizu, K.; Kimura, H.; Kimura, H.; Nishimura, M. Serum Alpha-1 Antitrypsin Levels and the Clinical Course of Chronic Obstructive Pulmonary Disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 2885–2893. [Google Scholar] [CrossRef] [Green Version]
- Gopal, P.; Reynaert, N.L.; Scheijen, J.L.I.M.; Schalkwijk, C.G.; Franssen, F.M.E.; Wouters, E.F.M.; Rutten, E.P.A. Association of plasma sRAGE, but not esRAGE with lung function impairment in COPD. Respir. Res. 2014, 15, 24. [Google Scholar] [CrossRef] [Green Version]
- Stoller, J.K.; Aboussouan, L.S. A review of alpha1-antitrypsin deficiency. Am. J. Respir. Crit. Care. Med. 2012, 185, 246–259. [Google Scholar] [CrossRef]
- Bowler, R.P.; Barnes, P.J.; Crapo, J.D. The role of oxidative stress in chronic obstructive pulmonary disease. COPD 2004, 1, 255–277. [Google Scholar] [CrossRef]
- Zemans, R.L.; Jacobson, S.; Keene, J.; Kechris, K.; Miller, B.E.; Tal-Singer, R.; Bowler, R.P. Multiple biomarkers predict disease severity, progression and mortality in COPD. Respir. Res. 2017, 18, 117. [Google Scholar] [CrossRef] [Green Version]
- Winterbach, W.; van Mieghem, P.; Reinders, M.; Wang, H.; de Ridder, D. Topology of molecular interaction networks. BMC Syst. Biol. 2013, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Gao, L. Biological network analysis: insights into structure and functions. Brief Funct. Genomics. 2012, 11, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawe, J.S.; Theis, F.J.; Heinig, M. Inferring Interaction Networks From Multi-Omics Data. Front. Genet. 2019, 10, 535. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014, 15, 34–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, E.A.; Hersh, C.P.; Castaldi, P.J.; DeMeo, D.L.; Silverman, E.K.; Crapo, J.D.; Bowler, R.P. Omics and the Search for Blood Biomarkers in Chronic Obstructive Pulmonary Disease. Insights from COPDGene. Am. J. Respir. Cell. Mol. Biol. 2019, 61, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.J.; Zhuang, Y.; Russell, P.H.; Hobbs, B.D.; Parker, M.M.; Castaldi, P.J.; Rudra, P.; Vestal, B.; Hersh, C.P.; Saba, L.M.; et al. Unsupervised discovery of phenotype-specific multi-omics networks. Bioinformatics 2019, 35, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Piazza, I.; Kochanowski, K.; Cappelletti, V.; Fuhrer, T.; Noor, E.; Sauer, U.; Picotti, P. A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication. Cell 2018, 172, 358–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Zhang, Q.; Zhou, Y.; Yu, S.; Hong, L.; Zhao, S.; Yang, J.; Wan, H.; Xu, G.; Zhang, Y.; et al. Integration of Proteomics and Metabolomics Revealed Metabolite-Protein Networks in ACTH-Secreting Pituitary Adenoma. Front. Endocrinol. 2018, 9, 678. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, W.; Feng, Y.; Guo, S.; Zhao, X.; Wang, Y.; He, Y.; He, W.; Chen, L. Prioritizing chronic obstructive pulmonary disease (COPD) candidate genes in COPD-related networks. Oncotarget 2017, 8, 103375–103384. [Google Scholar] [CrossRef] [Green Version]
- Bradford, E.; Jacobson, S.; Varasteh, J.; Comellas, A.P.; Woodruff, P.; O’Neal, W.; DeMeo, D.L.; Li, X.; Kim, V.; Cho, M.; et al. The value of blood cytokines and chemokines in assessing COPD. Respir. Res. 2017, 18, 180. [Google Scholar] [CrossRef] [Green Version]
- Bowler, R.P.; Bahr, T.M.; Hughes, G.; Lutz, S.; Kim, Y.; Coldren, C.D.; Reisdorph, N.; Kechris, K.J. Integrative omics approach identifies interleukin-16 as a biomarker of emphysema. OMICS 2013, 17, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Carolan, B.J.; Kim, Y.; Williams, A.A.; Kechris, K.; Lutz, S.; Reisdorph, N.; Bowler, R.P. The association of adiponectin with computed tomography phenotypes in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 2013, 188, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Judge, E.P.; Fabre, A.; Adamali, H.I.; Egan, J.I. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 40, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Brekke, P.H.; Omland, T.; Holmedal, S.H.; Smith, P.; Søyseth, V. Troponin T elevation and long-term mortality after chronic obstructive pulmonary disease exacerbation. Eur. Respir. J. 2008, 31, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Dempsie, Y.; Nilsen, M.; White, K.; Mair, K.M.; Loughlin, L.; Ambartsumian, N.; Rabinovitch, M.; MacLean, M.R. Development of pulmonary arterial hypertension in mice over-expressing S100A4/Mts1 is specific to females. Respir. Res. 2011, 12, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimann, S.; Fink, L.; Wilhelm, J.; Hoffmann, J.; Bednorz, M.; Seimetz, M.; Dessureault, I.; Troesser, R.; Ghanim, B.; Klepetko, W.; et al. Increased S100A4 expression in the vasculature of human COPD lungs and murine model of smoke-induced emphysema. Respir. Res. 2015. 16, 127. [CrossRef] [Green Version]
- Hattori, K.; Ishii, T.; Motegi, T.; Kusunoki, Y.; Gemma, A.; Kida, K. Relationship between serum cardiac troponin T level and cardiopulmonary function in stable chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2015, 10, 309–320. [Google Scholar] [PubMed] [Green Version]
- Gang, T.B.; Hammond, D.J., Jr.; Singh, S.K.; Ferguson, D.A., Jr.; Mishara, V.K.; Agrawal, A. The phosphocholine-binding pocket on C-reactive protein is necessary for initial protection of mice against pneumococcal infection. J. Biol. Chem. 2012, 287, 43116–43125. [Google Scholar] [CrossRef] [Green Version]
- Bernhard, W. Lung surfactant: Function and composition in the context of development and respiratory physiology. Ann. Anat. 2016, 208, 146–150. [Google Scholar] [CrossRef]
- Rahman, I.; Gilmour, P.S.; Jimenez, L.A.; Biswas, S.K.; Antonicelli, K.; Aruoma, O.I. Ergothioneine inhibits oxidative stress- and TNF-alpha-induced NF-kappa B activation and interleukin-8 release in alveolar epithelial cells. Biochem. Biophys. Res. Commun. 2003, 302, 860–864. [Google Scholar] [CrossRef]
- Adamson, R.; Swenson, E.R. Acetazolamide Use in Severe Chronic Obstructive Pulmonary Disease. Pros and Cons. Ann. Am. Thorac. Soc. 2017, 14, 1086–1093. [Google Scholar]
- Vallath, S.; Hynds, R.E.; Succony, L.; Janes, S.M.; Giangreco, A. Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. Eur. Respir. J. 2014, 44, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehkhoda, F.; Lee, C.M.M.; Medina, J.; Brooks, A.J. The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. 2018, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Barreiro, E.; Jaitovich, A. Muscle atrophy in chronic obstructive pulmonary disease: molecular basis and potential therapeutic targets. J. Thorac. Dis. 2018, 10, S1415–S1424. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Menon, R.K.; Chohen, P.; Hwang, D.; Clemens, T.; DiGirolamo, D.J.; Kopchick, J.J.; Le Roith, D.; Trucco, M.; Sperling, M.A. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J. Biol. Chem. 2009, 284, 19937–19944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jara, A.; Liu, X.; Sim, D.; Benner, C.M.; Duran-Ortiz, S.; Qian, Y.; List, E.O.; Berryman, D.E.; Kim, J.K.; Kopchick, J.J. Cardiac-Specific Disruption of GH Receptor Alters Glucose Homeostasis While Maintaining Normal Cardiac Performance in Adult Male Mice. Endocrinology 2016, 157, 1929–1941. [Google Scholar] [CrossRef] [PubMed]
- Coschigano, K.T.; Holland, A.N.; Riders, M.E.; List, E.O.; Flyvbjerg, A.; Kopchick, J.J. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology 2003, 144, 3799–3810. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Bennett, S.A.; Ingram, R.L.; Sonntag, W.E. Decreases in growth hormone receptor signal transduction contribute to the decline in insulin-like growth factor I gene expression with age. Endocrinology 1995, 136, 4551–4557. [Google Scholar] [CrossRef] [Green Version]
- Duran-Ortiz, S.; Noboa, V.; Kopchick, J.J. Tissue-specific disruption of the growth hormone receptor (GHR) in mice: An update. Growth Horm. IGF Res. 2019, 51, 1–5. [Google Scholar] [CrossRef]
- Jonker, R.; Deutz, N.E.P.; Erbland, N.L.; Anderson, P.J.; Engelen, M.P.K.J. Effectiveness of essential amino acid supplementation in stimulating whole body net protein anabolism is comparable between COPD patients and healthy older adults. Metabolism 2017, 69, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Makita, H.; Östling, J.; Thomsen, L.H.; Konno, S.; Nagai, K.; Shimizu, K.; Pederson, J.H.; Ashraf, H.; Bruijinzeel, P.L.B.; et al. Lower leptin/adiponectin ratio and risk of rapid lung function decline in chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2014, 11, 1511–1519. [Google Scholar] [CrossRef]
- Leivo-Korpela, S.; Lehtimäki, L.; Vuolteenaho, K.; Nieminen, R.; Kööbi, L.; Järvenpää, R.; Kankaanranta, H.; Saarelainen, S.; Moilanen, E. Adiponectin is associated with dynamic hyperinflation and a favourable response to inhaled glucocorticoids in patients with COPD. Respir. Med. 2014, 108, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.; Pham, A.; Cho, J.; Rosenthal, P.; Broide, D.H. Adiponectin-deficient mice are protected against tobacco-induced inflammation and increased emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, 834–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schols, A.M.W.J.; Creutzberg, E.C.; Buurman, W.A.; Campfield, L.A.; Saris, W.H.M.; Wouters, E.F.M. Plasma leptin is related to proinflammatory status and dietary intake in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care. Med. 1999, 160, 1220–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Gordon, E.M.; Figueroa, D.M.; Barochia, A.V.; Levine, S.J. Emerging Roles of Apolipoprotein E and Apolipoprotein A-I in the Pathogenesis and Treatment of Lung Disease. Am. J. Respir. Cell. Mol. Biol. 2016, 55, 59–169. [Google Scholar] [CrossRef] [Green Version]
- Yau, S.W.; Azar, W.J.; Sabin, M.A.; Werther, G.A.; Russo, V.C. IGFBP-2 - taking the lead in growth, metabolism and cancer. J. Cell. Commun. Signal. 2015, 9, 125–142. [Google Scholar] [CrossRef] [Green Version]
- Regan, E.A.; Hokanson, J.E.; Murphy, J.R.; Make, B.; Lynch, D.A.; Beaty, T.H.; Curran-Everett, D.; Silverman, E.K.; Crapo, J.D. Genetic epidemiology of COPD (COPDGene) study design. COPD 2010, 7, 32–43. [Google Scholar] [CrossRef]
- Li, K.; Gao, L.; Pan, Z.; Jia, X.; Yan, Y.; Min, X.; Huang, K.; Jiang, T. Influence of Emphysema and Air Trapping Heterogeneity on Pulmonary Function in Patients with COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 2863–2872. [Google Scholar] [CrossRef] [Green Version]
- Hankinson, J.L.; Odencrantz, J.R.; Fedan, K.B. Spirometric reference values from a sample of the general U.S. population. Am. J. Respir. Crit. Care. Med. 1999, 159, 179–187. [Google Scholar] [CrossRef]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.; Carter, J.; Cunningham, V.; Dalby, A.; Eaton, B.; et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef] [Green Version]
- Bijlsma, S.; Bobeldijk, I.; Verheij, E.R.; Ramaker, R.; Kochhar, S.; Macdonald, I.A.; van Ommen, B.; Smilde, A.K. Large-scale human metabolomics studies: a strategy for data (pre-) processing and validation. Anal. Chem. 2006, 78, 567–574. [Google Scholar] [CrossRef]
- Impute: Impute: Imputation for Microarray Data (Version 1.56.0.). Available online: http://bioconductor.statistik.tu-dortmund.de/packages/3.8/bioc/html/impute.html (accessed on 11 November 2018).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, D.; Schwikowski, B.; Ideker, T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G., Jr. Simultaneous Statistical Inference; Springer New York: New York, NY, USA, 1981; pp. 37–108. [Google Scholar]
- Yandall, B.S. Practical Data Analysis for Designed Experiments; CRC Press: Boca Raton, FL, USA, 1997; pp. 1–440. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Clinical Variables | Control (n = 426) | COPD (n = 478) | PRISm (n = 92) | Missing Spirometry (n = 12) | Whole Cohort (n = 1008) |
|---|---|---|---|---|---|
| Sex, % women | 53.3 | 42.7 | 63 | 50 | 49.1 |
| Race, % white | 88.9 | 94.8 | 91.3 | 100 | 92.1 |
| Former Smoker, % | 73.7 | 77.6 | 70.7 | 66.7 | 75.2 |
| Age (yr) | 64.6 (58.3–71.5) | 71.1 (64.9–76.6) | 67.3 (60.9–72.9) | 72 (68.2–75.3) | 68 (61–74.6) |
| Body mass index (kg/m2) | 28.4 (25.3–32.1) | 27.4 (23.7–31.7) | 30.8 (27.4–37.4) | 27.8 (25.2–30.2) | 28.1 (24.7–32.2) |
| Heart disease comorbidity (%) | 30.3 | 50.2 | 38 | 50 | 40.7 |
| FEV1% predicted | 106.8 (11.7) | 58(23.5) | 70 (7.7) | NA | 77.4 (26.6) * |
| Percent emphysema (% LAA < −950 HU) | 2.2 (2.6) | 12.8 (12.3) | 1.5 (2.6) | 9.2 (11.7) | 7.1 (10.1) ** |
| -Omics Type | Network Node | Correlation to FEV1 (%) |
|---|---|---|
| Proteins | Troponin T | −0.254 |
| Protein S100-A4 | 0.187 | |
| Alpha-(1,3)-fucosyltransferase 5 | −0.175 | |
| Carbonic anhydrase 6 | 0.171 | |
| RGMA | 0.152 | |
| Epidermal growth factor receptor | 0.151 | |
| Hemojuvelin | 0.146 | |
| C-reactive protein | −0.144 | |
| Macrophage mannose receptor 1 | −0.144 | |
| Kallistatin | 0.141 | |
| Angiopoietin-2 | −0.140 | |
| RBP | 0.138 | |
| Complement component C9 | −0.135 | |
| Metabolites | Phosphocholine | 0.250 |
| Ergothioneine | 0.220 | |
| 5-hydroxyhexanoate | −0.213 | |
| Palmitoleoylcarnitine (C16:1) | −0.205 | |
| Myristoleoylcarnitine (C14:1) | −0.200 | |
| Cis-4-decenoylcarnitine (C10:1) | −0.199 | |
| (N(1) + N(8))-acetylspermidine | −0.184 |
| -Omics Type | Network Node | Correlation to Percent Emphysema |
|---|---|---|
| Proteins | Troponin T | 0.197 |
| Leptin | −0.169 | |
| Glucagon | −0.163 | |
| Growth hormone receptor | −0.161 | |
| Proto-oncogene tyrosine-protein kinase receptor Ret | −0.146 | |
| Chordin-like protein 1 | 0.143 | |
| Hemojuvelin | −0.142 | |
| Sex hormone-binding globulin | 0.139 | |
| Aminoacylase-1 | −0.138 | |
| Adiponectin | 0.137 | |
| Apolipoprotein E | −0.130 | |
| IGFBP-2 | 0.119 | |
| QORL1 | −0.106 | |
| Metabolites | 1-stearoyl-2-linoleoyl-GPI (18:0/18:2) | −0.209 |
| androsterone glucuronide | −0.206 | |
| 1-stearoyl-2-docosahexaenoyl-GPE (18:0/22:6) | −0.200 | |
| 1-palmitoyl-2-docosahexaenoyl-GPE (16:0/22:6) | −0.188 | |
| 1-palmitoyl-2-linoleoyl-GPI (16:0/18:2) | −0.187 | |
| 1-ribosyl-imidazoleacetate | −0.173 | |
| Valine | −0.168 | |
| palmitoyl-linoleoyl-glycerol (16:0/18:2) [2] | −0.166 | |
| 1-stearoyl-2-arachidonoyl-GPI (18:0/20:4) | −0.161 | |
| Glutamate | −0.151 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mastej, E.; Gillenwater, L.; Zhuang, Y.; Pratte, K.A.; Bowler, R.P.; Kechris, K. Identifying Protein–metabolite Networks Associated with COPD Phenotypes. Metabolites 2020, 10, 124. https://doi.org/10.3390/metabo10040124
Mastej E, Gillenwater L, Zhuang Y, Pratte KA, Bowler RP, Kechris K. Identifying Protein–metabolite Networks Associated with COPD Phenotypes. Metabolites. 2020; 10(4):124. https://doi.org/10.3390/metabo10040124
Chicago/Turabian StyleMastej, Emily, Lucas Gillenwater, Yonghua Zhuang, Katherine A. Pratte, Russell P. Bowler, and Katerina Kechris. 2020. "Identifying Protein–metabolite Networks Associated with COPD Phenotypes" Metabolites 10, no. 4: 124. https://doi.org/10.3390/metabo10040124
APA StyleMastej, E., Gillenwater, L., Zhuang, Y., Pratte, K. A., Bowler, R. P., & Kechris, K. (2020). Identifying Protein–metabolite Networks Associated with COPD Phenotypes. Metabolites, 10(4), 124. https://doi.org/10.3390/metabo10040124