Identifying Personalized Metabolic Signatures in Breast Cancer

 ,
, 
Abstract
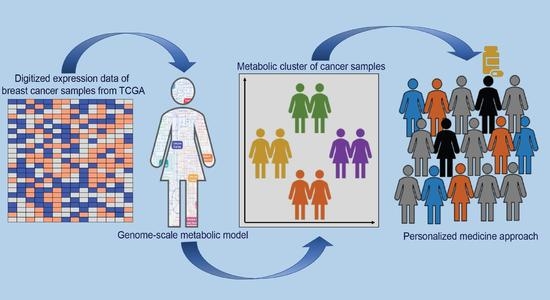
:
1. Introduction
2. Results
2.1. Understanding Metabolic Differences in Cancer Samples Using Personalized Metabolic Networks
2.2. Classifying Cancer Samples Based on Their Metabolic Profile
- (a)
- Class comparison: We compared the reaction fluxes for cancer and normal samples in the dataset and classified reactions in each context-specific network as active or inactive based on their flux measurement (described in the methods section). In order to identify active reactions in the context-specific networks, we used the information of reaction fluxes from all 1156 context-specific metabolic networks. If a reaction was present in the network, it was assigned a state of 1, while the remaining reactions were assigned a state of 0, indicating that they were absent in the context-specific metabolic network. Statistical analysis of active reactions in divergent networks identified 471 reactions (p-value < 0.05) that were significantly different in cancer versus normal. These reactions belonged to the following pathways: androgen and estrogen metabolism, bile acid synthesis, cholesterol metabolism, citric acid cycle, drug metabolism, eicosanoid metabolism, exchange reactions, fatty acid oxidation, glutathione metabolism, glycerophospholipid metabolism, glycolysis, steroid metabolism, transport, tyrosine metabolism, urea cycle, and vitamin metabolism. Supplementary Table S1 represents the list of subsystems that were enriched in cancer versus normal.
- (b)
- Class discovery: We used an unsupervised machine learning method to classify the cancer samples based on their metabolic state. Using K-means clustering on the simulated reaction fluxes, we obtained four distinct clusters of cancer samples (Figure 2c). The number of clusters was determined by the elbow method; see Supplementary Figure S2. The cancer clusters were then labeled from one to four, and normal tissue samples were assigned as cluster 0. We performed a detailed analysis of the four clusters to identify, if any, associations with standard clinical and pathological tumor characteristics. This analysis showed that the metabolic clusters were significantly associated with PAM50 molecular subtypes and estrogen receptor (ER) status (chi-squared p-value < 0.001), distinguishing the luminal A and B samples from basal-like samples, and also ER-positive and negative samples to a greater extent. Specifically, cluster two was enriched for luminal subtypes (luminal A and B) and predominantly accounted for ER-positive samples, while cluster three was enriched in basal-like and ER-negative tumors. (Figure 3 and Supplementary file 1). The metabolic clusters of tumor and normal samples were used for identifying important reactions and subsystems in these clusters.
2.3. Identifying Candidate Druggable Genes
3. Discussion
4. Materials and Methods
4.1. Expression Data and Divergence Analysis
4.2. Integration of Expression Data to Generate Personalized Metabolic Networks
4.3. Classification of Context-Specific Metabolic Networks into Metabolic Subgroups
4.4. Identifying Target Genes in the Context-Specific Networks
4.5. Metabolic Genes Divergent in the Expression and Methylation Space
4.6. Drug Target Identification for Genes Shortlisted from Metabolic Networks
4.7. Statistical Analysis
4.8. Software
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiang, A.C.; Massagué, J. Molecular basis of metastasis. N. Engl. J. Med. 2008, 359, 2814–2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhao, S. Metabolic changes in cancer: Beyond the Warburg effect. Acta Biochim. Biophys. Sin. 2013, 45, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinalankara, W.; Ke, Q.; Xu, Y.; Ji, L.; Pagane, N.; Lien, A.; Matam, T.; Fertig, E.J.; Price, N.D.; Younes, L.; et al. Digitizing omics profiles by divergence from a baseline. Proc. Natl. Acad. Sci. USA 2018, 115, 4545–4552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Lu, W.; Akutsu, T. Computational Methods for Modification of Metabolic Networks. Comput. Struct. Biotechnol. J. 2015, 13, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzaniga, P.; Damiani, C.; Besozzi, D.; Colombo, R.; Nobile, M.S.; Gaglio, D.; Pescini, D.; Molinari, S.; Mauri, G.; Alberghina, L.; et al. Computational strategies for a system-level understanding of metabolism. Metabolites 2014, 4, 1034–1087. [Google Scholar] [CrossRef]
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst. Biol. 2012, 6, 153. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.J.; Nielsen, J. Genome-scale metabolic models applied to human health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1393. [Google Scholar] [CrossRef]
- Raman, K.; Chandra, N. Flux balance analysis of biological systems: Applications and challenges. Brief. Bioinform. 2009, 10, 435–449. [Google Scholar] [CrossRef]
- Nilsson, A.; Nielsen, J. Genome scale metabolic modeling of cancer. Metab. Eng. 2017, 43, 103–112. [Google Scholar] [CrossRef]
- Zhang, C.; Aldrees, M.; Arif, M.; Li, X.; Mardinoglu, A.; Aziz, M.A. Elucidating the Reprograming of Colorectal Cancer Metabolism Using Genome-Scale Metabolic Modeling. Front. Oncol. 2019, 9, 681. [Google Scholar] [CrossRef] [Green Version]
- Jerby, L.; Ruppin, E. Predicting drug targets and biomarkers of cancer via genome-scale metabolic modeling. Clin. Cancer Res. 2012, 18, 5572–5584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yizhak, K.; Chaneton, B.; Gottlieb, E.; Ruppin, E. Modeling cancer metabolism on a genome scale. Mol. Syst. Biol. 2015, 11, 817. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar]
- Bao, X.; Anastasov, N.; Wang, Y.; Rosemann, M. A novel epigenetic signature for overall survival prediction in patients with breast cancer. J. Transl. Med. 2019, 17, 1–12. [Google Scholar] [CrossRef]
- de Almeida, B.P.; Apolónio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer 2019, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Hammond-Martel, I.; Pak, H.; Yu, H.; Rouget, R.; Horwitz, A.A.; Parvin, J.D.; Drobetsky, E.A.; Affar, E.B. PI 3 kinase related kinases-independent proteolysis of BRCA1 regulates Rad51 recruitment during genotoxic stress in human cells. PLoS ONE 2010, 5, e14027. [Google Scholar] [CrossRef]
- Verma, S.; Yeddula, N.; Soda, Y.; Zhu, Q.; Pao, G.; Moresco, J.; Diedrich, J.K.; Hong, A.; Plouffe, S.; Moroishi, T.; et al. BRCA1/BARD1-dependent ubiquitination of NF2 regulates Hippo-YAP1 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 7363–7370. [Google Scholar] [CrossRef] [Green Version]
- Guo, N.; Peng, Z. MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia Pac. J. Clin. Oncol. 2013, 9, 6–11. [Google Scholar] [CrossRef]
- Han, Y.H.; Moon, H.J.; You, B.R.; Park, W.H. The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol. Rep. 2009, 22, 215–221. [Google Scholar] [PubMed]
- Zhang, Y.; Yang, B.; Zhao, J.; Li, X.; Zhang, L.; Zhai, Z. Proteasome Inhibitor Carbobenzoxy-L-Leucyl-L-Leucyl-L-Leucinal (MG132) Enhances Therapeutic Effect of Paclitaxel on Breast Cancer by Inhibiting Nuclear Factor (NF)-κB Signaling. Med. Sci. Monit. 2018, 24, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, S.-C.; Kashida, Y.; Kulp, S.K.; Wang, D.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.-S. Sensitizing estrogen receptor-negative breast cancer cells to tamoxifen with OSU-03012, a novel celecoxib-derived phosphoinositide-dependent protein kinase-1/Akt signaling inhibitor. Mol. Cancer Ther. 2008, 7, 800–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. OSU-03012 in the treatment of glioblastoma. Mol. Pharmacol. 2006, 70, 437–439. [Google Scholar] [CrossRef]
- Wang, F.; Wang, L.; Zhao, Y.; Li, Y.; Ping, G.; Xiao, S.; Chen, K.; Zhu, W.; Gong, P.; Yang, J.; et al. A novel small-molecule activator of procaspase-3 induces apoptosis in cancer cells and reduces tumor growth in human breast, liver and gallbladder cancer xenografts. Mol. Oncol. 2014, 8, 1640–1652. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med. 2014, 11, 1–19. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.-O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. Plos Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Thiele, I. Computationally efficient flux variability analysis. BMC Bioinform. 2010, 11, 489. [Google Scholar] [CrossRef] [Green Version]
- MacQueen, J. Others Some methods for classification and analysis of multivariate observations. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability, Oakland, CA, USA, 21 June–18 July 1967; Volume 1, pp. 281–297. [Google Scholar]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, 6352. [Google Scholar] [CrossRef] [Green Version]
- Drug Download Page—Cancerrxgene—Genomics of Drug Sensitivity in Cancer. Available online: https://www.cancerrxgene.org/downloads/bulk_download (accessed on 24 February 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subsystem | Gene | Drug Target |
|---|---|---|
| Cholesterol metabolism | SOAT1 | FDA approved |
| Valine, leucine, and isoleucine metabolism | MUT | FDA approved |
| Citric acid cycle | SDHA, SDHB, SDHC, SDHD | FDA approved (SDHD), Potential drug target |
| C5-branched dibasic acid metabolism | SUCLA2, SUCLG1, SUCLG2 | Potential drug target |
| Lysine metabolism | DLD, DLST | Potential drug target |
| Oxidative phosphorylation | ATP5 family, COX family, UQCR family, CYC1, CYTB | Potential drug target |
| Pyrimidine synthesis | UPRT | |
| Sphingolipid metabolism | SGMS1 | |
| Transport, mitochondrial | SLC25A10 | |
| Glycerophospholipid metabolism | CEPT1, PCYT2, PDHX |
| Drug | Brand Name | Target | #Significant Samples (out of 1156) | Cohort |
|---|---|---|---|---|
| MG-132 | Proteasome | 599 | BRCA | |
| OSU-03012 | PDPK1 (PDK1) | 474 | All cell lines | |
| PAC-1 | CASP3 agonist | 94 | All cell lines | |
| GSK-1904529A | IGF1R | 89 | All cell lines | |
| PF-562271 | FAK | 31 | All cell lines | |
| QS11 | ARFGAP | 28 | All cell lines | |
| Trametinib | Mekinist | MAP2K1 (MEK1), MAP2K2 (MEK2) | 28 | All cell lines |
| XMD11-85h | BRSK2, FLT4, MARK4, PRKCD, RET, SPRK1 | 23 | All cell lines | |
| (5Z)-7-Oxozeaenol | MAP3K7 (TAK1) | 14 | All cell lines | |
| GSK-650394 | SGK3 | 12 | All cell lines | |
| Tipifarnib | Zarnestra, IND58359, R115777 | Farnesyl-transferase (FNTA) | 12 | All cell lines |
| Vinorelbine | Navelbine | Microtubules | 8 | All cell lines |
| 5-Fluorouracil | DNA antimetabolite | 5 | All cell lines |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baloni, P.; Dinalankara, W.; Earls, J.C.; Knijnenburg, T.A.; Geman, D.; Marchionni, L.; Price, N.D. Identifying Personalized Metabolic Signatures in Breast Cancer. Metabolites 2021, 11, 20. https://doi.org/10.3390/metabo11010020
Baloni P, Dinalankara W, Earls JC, Knijnenburg TA, Geman D, Marchionni L, Price ND. Identifying Personalized Metabolic Signatures in Breast Cancer. Metabolites. 2021; 11(1):20. https://doi.org/10.3390/metabo11010020
Chicago/Turabian StyleBaloni, Priyanka, Wikum Dinalankara, John C. Earls, Theo A. Knijnenburg, Donald Geman, Luigi Marchionni, and Nathan D. Price. 2021. "Identifying Personalized Metabolic Signatures in Breast Cancer" Metabolites 11, no. 1: 20. https://doi.org/10.3390/metabo11010020
APA StyleBaloni, P., Dinalankara, W., Earls, J. C., Knijnenburg, T. A., Geman, D., Marchionni, L., & Price, N. D. (2021). Identifying Personalized Metabolic Signatures in Breast Cancer. Metabolites, 11(1), 20. https://doi.org/10.3390/metabo11010020