Metabolic Detoxification of 2-Oxobutyrate by Remodeling Escherichia coli Acetate Bypass



 and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Metabolic Toxicity Detection of 2-OBA Using an l-Threonine-Producing E. coli Strain
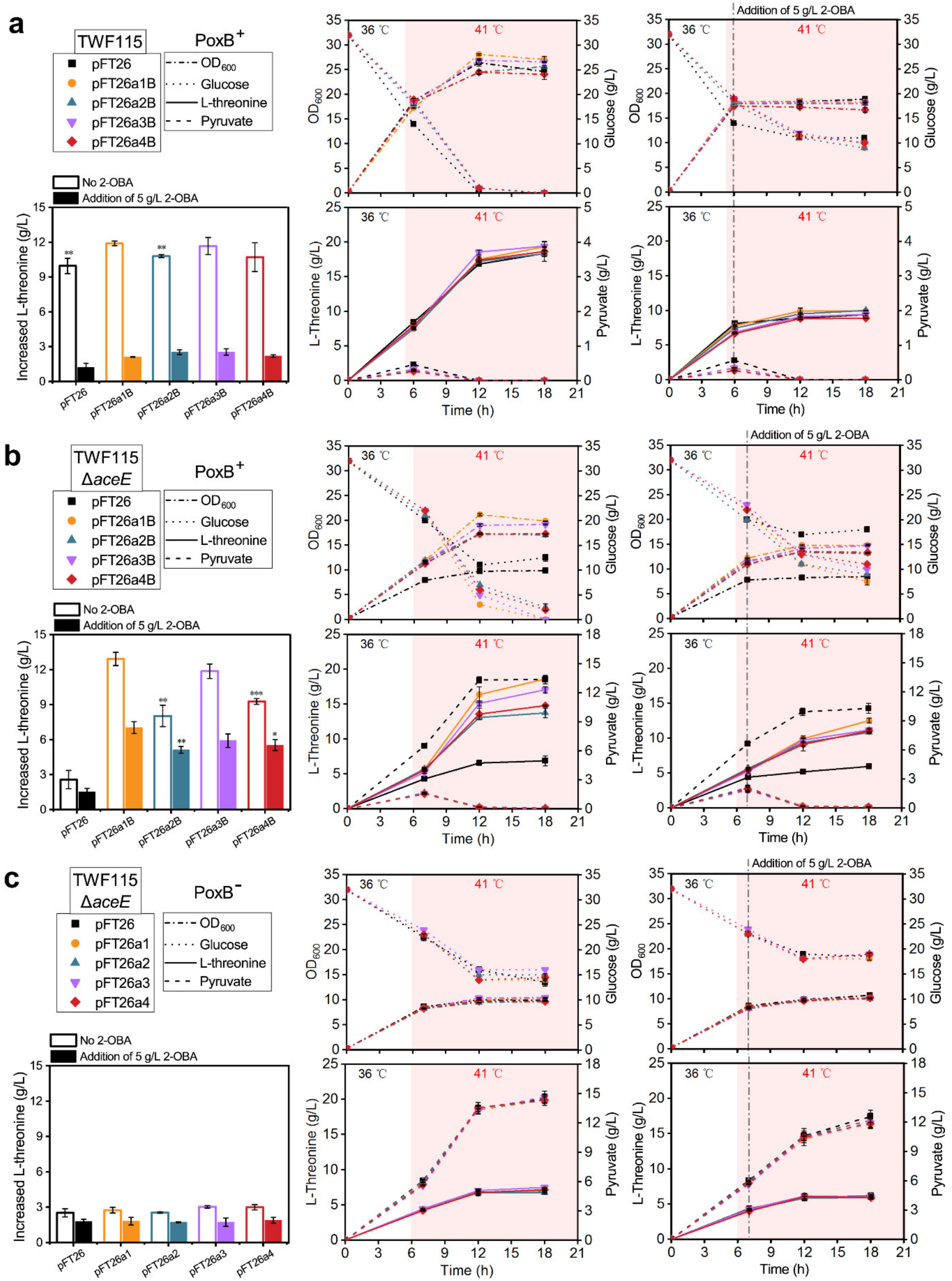
2.2. Alleviation of the Metabolic Toxicity of 2-OBA in the Engineered E. coli Strains
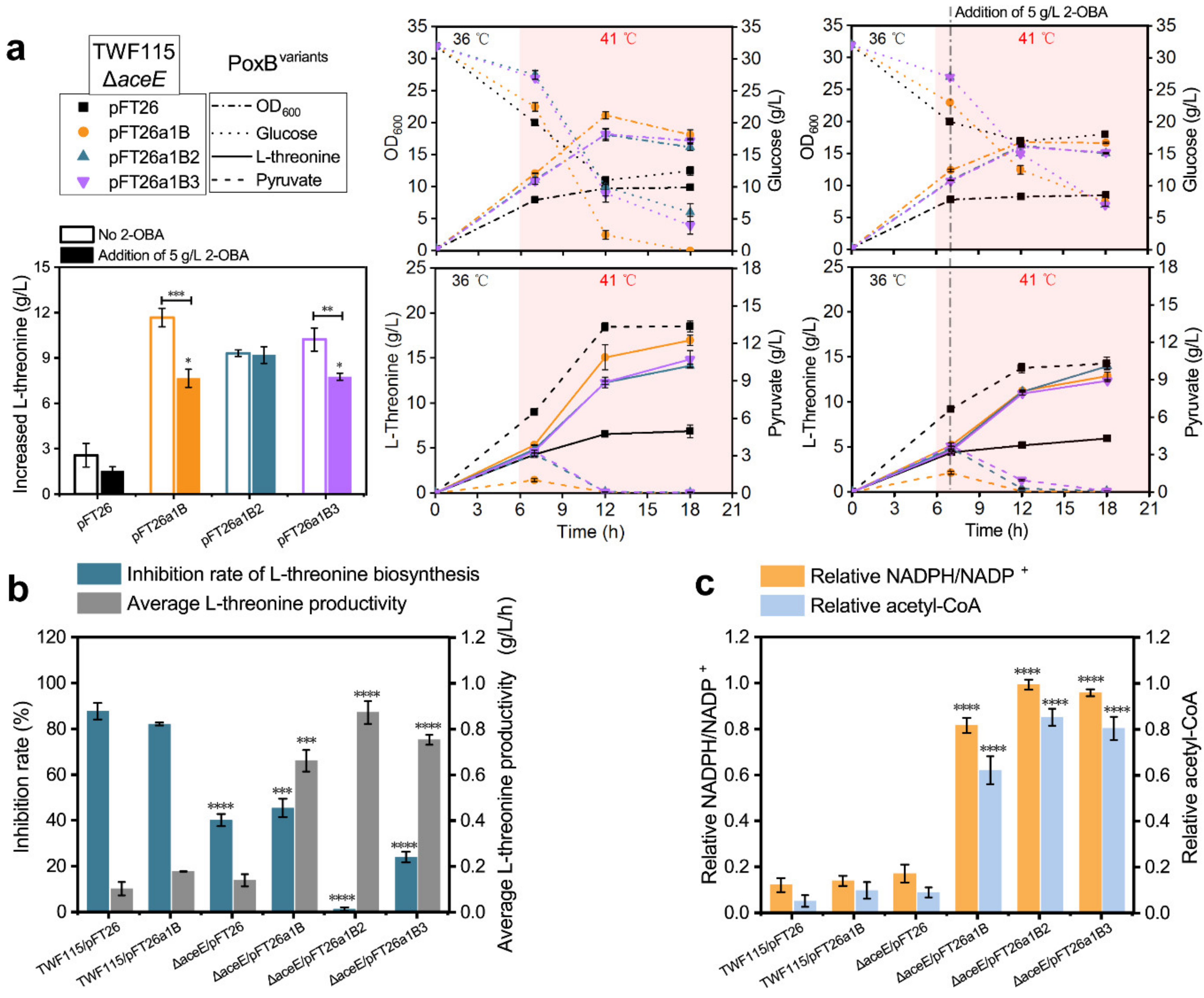
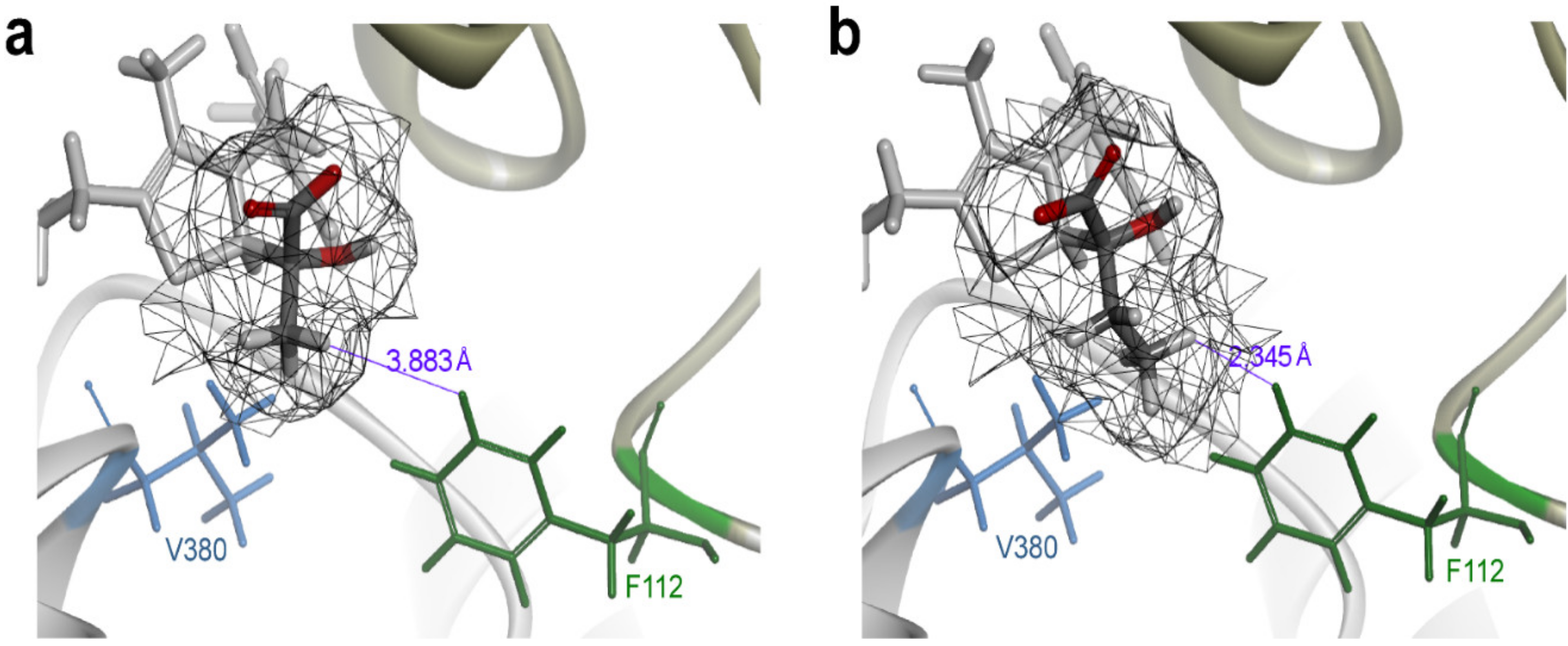
2.3. Directed Evolution of PoxB to Improve its Selectivity toward Pyruvate
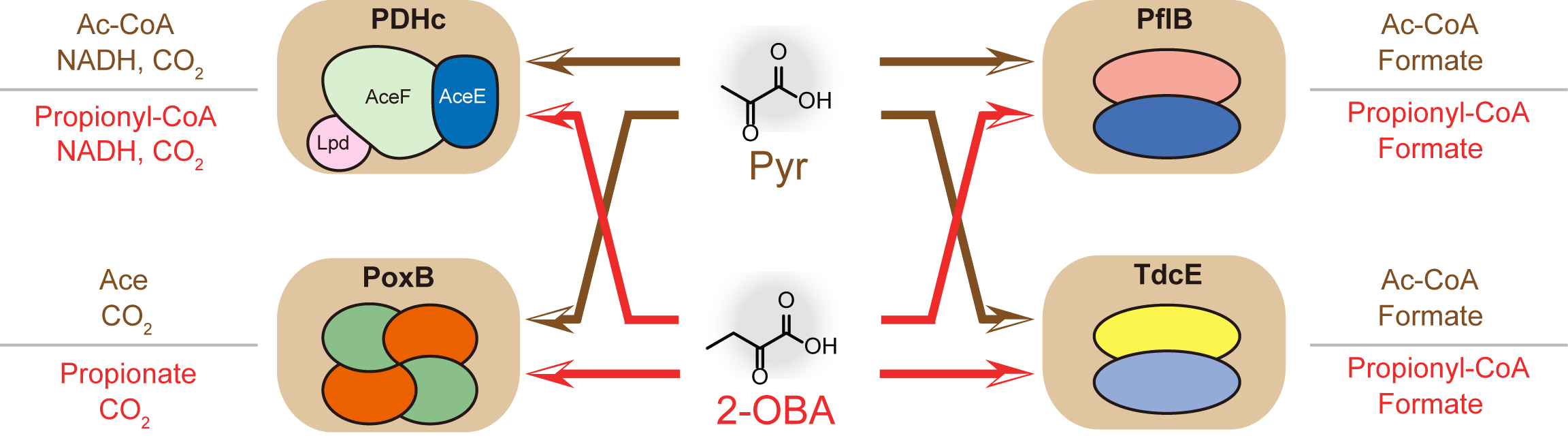
2.4. Detoxification of 2-OBA for l-Threonine Biosynthesis by Reconstructing the Acetate Bypass in E. coli
3. Materials and Methods
3.1. Bacterial Strains and Plasmids
3.2. Plasmids and DNA Manipulation
3.3. Culture and Fermentation Conditions
3.4. Molecular Docking Simulation
3.5. Library Screening of PoxB Variants
3.6. PoxB Activity Assays
3.7. Extracellular Metabolite Analysis
3.8. Intracellular NADPH and Acetyl-CoA Quantification
3.9. Statistical Analysis and Reproducibility
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, C.; Kim, I.; Park, C. Glyoxal detoxification in Escherichia coli K-12 by NADPH dependent aldo-keto reductases. J. Microbiol. 2013, 51, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, G.P.; Munro, A.W.; Douglas, R.M.; McLaggan, D.; Booth, I.R. Activation of potassium channels during metabolite detoxification in Escherichia coli. Mol. Microbiol. 1993, 9, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cavka, A.; Jönsson, L.J. Detoxification of lignocellulosic hydrolysates using sodium borohydride. Bioresour. Technol. 2013, 136, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jönsson, L.J.; Alriksson, B.; Nilvebrant, N.O. Bioconversion of lignocellulose: Inhibitors and detoxification. Biotechnol. Biofuels 2013, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.; Squillace, P.; Gilcrease, P.C.; Menkhaus, T.J. Detoxification of a lignocellulosic biomass slurry by soluble polyelectrolyte adsorption for improved fermentation efficiency. Biotechnol. Bioeng. 2011, 108, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, J.; Shin, H.D.; Liu, L.; Du, G.; Chen, J. Biotechnological production of alpha-keto acids: Current status and perspectives. Bioresour. Technol. 2016, 219, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Ginos, J.Z.; Meister, A. Synthesis and properties of the. alpha.-keto acids. Chem. Rev. 1983, 83, 321–358. [Google Scholar] [CrossRef]
- LaRossa, R.A.; Van Dyk, T.K.; Smulski, D.R. Toxic accumulation of alpha-ketobutyrate caused by inhibition of the branched-chain amino acid biosynthetic enzyme acetolactate synthase in Salmonella typhimurium. J. Bacteriol. 1987, 169, 1372–1378. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Oh, J.E.; Lee, K.H.; Kim, J.Y.; Lee, S.Y. Rational design of Escherichia coli for L-isoleucine production. ACS Synth. Biol. 2012, 1, 532–540. [Google Scholar] [CrossRef]
- Yin, L.; Hu, X.; Xu, D.; Ning, J.; Chen, J.; Wang, X. Co-expression of feedback-resistant threonine dehydratase and acetohydroxy acid synthase increase L-isoleucine production in Corynebacterium glutamicum. Metab. Eng. 2012, 14, 542–550. [Google Scholar] [CrossRef]
- Xu, J.M.; Li, J.Q.; Zhang, B.; Liu, Z.Q.; Zheng, Y.G. Fermentative production of the unnatural amino acid L-2-aminobutyric acid based on metabolic engineering. Microb. Cell Fact. 2019, 18, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsumi, S.; Hanai, T.; Liao, J.C. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 2008, 451, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Lan, E.I.; Liao, J.C. Microbial synthesis of n-butanol, isobutanol, and other higher alcohols from diverse resources. Bioresour. Technol. 2013, 135, 339–349. [Google Scholar] [CrossRef] [PubMed]
- El-Dalatony, M.M.; Saha, S.; Govindwar, S.P.; Abou-Shanab, R.A.I.; Jeon, B.H. Biological Conversion of Amino Acids to Higher Alcohols. Trends Biotechnol. 2019, 37, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Furuyoshi, S.; Nawa, Y.; Kawabata, N. Microbial production of 2-oxobutyric acid from crotonic acid. Agric. Biol. Chem. 1991, 55, 123–128. [Google Scholar]
- Wang, Y.; Feng, Y.; Xue, S. Highly efficient production of 2-ketobutyrate from 2-hydroxybutyrate by resting cells of Pseudomonas stutzeri DEH130. Biocatal. Biotransfor. 2016, 34, 189–193. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, C.; Che, B.; Ma, C.; Zheng, Z.; Qin, T.; Xu, P. Efficient bioconversion of l-threonine to 2-oxobutyrate using whole cells of Pseudomonas stutzeri SDM. Bioresour. Technol. 2012, 110, 719–722. [Google Scholar] [CrossRef]
- Zhang, C.; Qi, J.; Li, Y.; Fan, X.; Xu, Q.; Chen, N.; Xie, X. Production of α-ketobutyrate using engineered Escherichia coli via temperature shift. Biotechnol. Bioeng. 2016, 113, 2054–2059. [Google Scholar] [CrossRef]
- Barak, Z.; Chipman, D.M.; Gollop, N. Physiological implications of the specificity of acetohydroxy acid synthase isozymes of enteric bacteria. J. Bacteriol. 1987, 169, 3750–3756. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Y.; Wang, X. Acetohydroxyacid synthases: Evolution, structure, and function. Appl. Microbiol. Biotechnol. 2016, 100, 8633–8649. [Google Scholar] [CrossRef]
- Stephens, P.E.; Darlison, M.G.; Lewis, H.M.; Guest, J.R. The pyruvate dehydrogenase complex of Escherichia coli K12. Nucleotide sequence encoding the pyruvate dehydrogenase component. Eur. J. Biochem. 1983, 133, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Arjunan, P.; Nemeria, N.; Brunskill, A.; Chandrasekhar, K.; Sax, M.; Yan, Y.; Jordan, F.; Guest, J.R.; Furey, W. Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 A resolution. Biochemistry 2002, 41, 5213–5221. [Google Scholar] [CrossRef]
- Sitte, A. Catalysis at the Interface-Elucidation of the Activation Process and Coupling of Catalysis and Compartmentalization of the Peripheral Membrane Protein Pyruvate Oxidase from Escherichia coli. Ph.D. Thesis, Niedersächsische Staats-und Universitätsbibliothek Göttingen, Göttingen, Germany, 2013. [Google Scholar]
- Hesslinger, C.; Fairhurst, S.A.; Sawers, G. Novel keto acid formate-lyase and propionate kinase enzymes are components of an anaerobic pathway in Escherichia coli that degrades l-threonine to propionate. Mol. Microbiol. 1998, 27, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Tanaka, T.; Kondo, A. Engineering metabolic pathways in Escherichia coli for constructing a “microbial chassis” for biochemical production. Bioresour. Technol. 2017, 245, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Satowa, D.; Fujiwara, R.; Uchio, S.; Nakano, M.; Otomo, C.; Hirata, Y.; Matsumoto, T.; Noda, S.; Tanaka, T.; Kondo, A. Metabolic engineering of E. coli for improving mevalonate production to promote NADPH regeneration and enhance acetyl-CoA supply. Biotechnol. Bioeng. 2020, 117, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, W.; Fang, Y.; Yang, J.; Zhan, J.; Chen, S.; Wang, X. Increasing l-threonine production in Escherichia coli by overexpressing the gene cluster phaCAB. J. Ind. Microbiol. Biotechnol. 2019, 46, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, W.; Fang, Y.; Zhang, H.; Liang, H.; Li, Y.; Wang, X. Truncating the Structure of Lipopolysaccharide in Escherichia coli Can Effectively Improve Poly-3-hydroxybutyrate Production. ACS Synth. Biol. 2020, 9, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, Y.; Kubo, T.; Soma, Y.; Saruta, F.; Hanai, T. Enhancement of acetyl-CoA flux for photosynthetic chemical production by pyruvate dehydrogenase complex overexpression in Synechococcus elongatus PCC 7942. Metab. Eng. 2020, 57, 23–30. [Google Scholar] [CrossRef]
- Wang, L.; Zong, Z.; Liu, Y.; Zheng, M.; Li, D.; Wang, C.; Zheng, F.; Madzak, C.; Liu, Z. Metabolic engineering of Yarrowia lipolytica for the biosynthesis of crotonic acid. Bioresour. Technol. 2019, 287, 121484. [Google Scholar] [CrossRef]
- Yu, Y.; Shao, M.; Li, D.; Fan, F.; Xu, H.; Lu, F.; Bi, C.; Zhu, X.; Zhang, X. Construction of a carbon-conserving pathway for glycolate production by synergetic utilization of acetate and glucose in Escherichia coli. Metab. Eng. 2020, 61, 152–159. [Google Scholar] [CrossRef]
- Soma, Y.; Yamaji, T.; Matsuda, F.; Hanai, T. Synthetic metabolic bypass for a metabolic toggle switch enhances acetyl-CoA supply for isopropanol production by Escherichia coli. J. Biosci. Bioeng. 2017, 123, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Weidner, A.; Pech, A.; Stubbs, M.T.; Tittmann, K. Structural basis for membrane binding and catalytic activation of the peripheral membrane enzyme pyruvate oxidase from Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 17390–17395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.Y.; Cronan, J.E., Jr. Conversion of Escherichia coli pyruvate oxidase to an ‘alpha-ketobutyrate oxidase’. Biochem. J. 2000, 352 Pt 3, 717–724. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, J.; Ma, W.; Yang, J.; Zhang, H.; Zhao, L.; Chen, S.; Zhang, S.; Hu, X.; Li, Y.; et al. Rebalancing microbial carbon distribution for l-threonine maximization using a thermal switch system. Metab. Eng. 2020, 61, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Xiong, H.; Xie, X.; Chen, N.; Zhao, G.; Caiyin, Q.; Zhu, H.; Qiao, J. Two-stage carbon distribution and cofactor generation for improving l-threonine production of Escherichia coli. Biotechnol. Bioeng. 2019, 116, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wei, H.; Wang, T.; Xu, Q.; Zhang, C.; Fan, X.; Ma, Q.; Chen, N.; Xie, X. Current status on metabolic engineering for the production of l-aspartate family amino acids and derivatives. Bioresour. Technol. 2017, 245, 1588–1602. [Google Scholar] [CrossRef]
- Starai, V.J.; Gardner, J.G.; Escalante-Semerena, J.C. Residue Leu-641 of Acetyl-CoA synthetase is critical for the acetylation of residue Lys-609 by the Protein acetyltransferase enzyme of Salmonella enterica. J. Biol. Chem. 2005, 280, 26200–26205. [Google Scholar] [CrossRef] [Green Version]
- Novak, K.; Flöckner, L.; Erian, A.M.; Freitag, P.; Herwig, C.; Pflügl, S. Characterizing the effect of expression of an acetyl-CoA synthetase insensitive to acetylation on co-utilization of glucose and acetate in batch and continuous cultures of E. coli W. Microb. Cell Fact. 2018, 17, 109. [Google Scholar] [CrossRef]
- Akita, H.; Nakashima, N.; Hoshino, T. Pyruvate production using engineered Escherichia coli. AMB Express 2016, 6, 94. [Google Scholar] [CrossRef] [Green Version]
- Causey, T.B.; Shanmugam, K.T.; Yomano, L.P.; Ingram, L.O. Engineering Escherichia coli for efficient conversion of glucose to pyruvate. Proc. Natl. Acad. Sci. USA 2004, 101, 2235–2240. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hamid, A.M.; Attwood, M.M.; Guest, J.R. Pyruvate oxidase contributes to the aerobic growth efficiency of Escherichia coli. Microbiology 2001, 147, 1483–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Chen, L.; Hei, M.; Liu, T.; Feng, Y.; Yang, G.Y. Structure-guided reshaping of the acyl binding pocket of ‘TesA thioesterase enhances octanoic acid production in E. coli. Metab. Eng. 2020, 61, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.Y.; Cronan, J.E., Jr. Mapping nonselectable genes of Escherichia coli by using transposon Tn10: Location of a gene affecting pyruvate oxidase. J. Bacteriol. 1982, 151, 1279–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Fang, Y.; Wang, X.; Zhao, L.; Wang, J.; Li, Y. Increasing l-threonine production in Escherichia coli by engineering the glyoxylate shunt and the l-threonine biosynthesis pathway. Appl. Microbiol. Biotechnol. 2018, 102, 5505–5518. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Additional 2-OBA (g/L) | 0 | 1.0 | 2.5 | 5.0 | 10.0 |
|---|---|---|---|---|---|
| Increased l-threonine (g/L) | 10.95 ± 0.66 | 9.32 ± 0.13 | 6.18 ± 0.18 | 1.23 ± 0.29 | 1.24 ± 0.32 |
| Inhibition rate of l-threonine biosynthesis (%) | 0 | 14.86 ± 1.15 | 43.53 ± 1.67 | 88.79 ± 2.26 | 88.71 ± 2.94 |
| Enzyme | Relative Activity (%) | Activity Ratio (Pyr/2-OBA) | |
|---|---|---|---|
| Pyruvate | 2-OBA | ||
| None | 0 | 0 | - |
| WT | 100 ± 2.51 | 16.06 ± 0.07 | 6.23 |
| V380C | 33.53 ± 2.35 | 9.37 ± 0.21 | 3.58 |
| V380I | 18.82 ± 0.52 | 4.24 ± 0.26 | 4.44 |
| V380T | 10.33 ± 0.99 | 2.80 ± 0.12 | 3.69 |
| F112A | 5.05 ± 0.12 | 1.66 ± 0.06 | 3.04 |
| F112L | 8.93 ± 0.25 | 8.57 ± 0.25 | 1.04 |
| F112T | 1.69 ± 0.36 | 0.51 ± 0.12 | 3.31 |
| F112W | 18.41 ± 0.16 | 0.92 ± 0.07 | 20.01 |
| F112W/L253F | 26.88 ± 0.36 | 3.89 ± 0.12 | 6.91 |
| Primer Name | Sequence (5′-3′) |
|---|---|
| poxB-PR-F | ATGGTTGCATGAATTCGATCCCTCCGTCAGATGAACTAAAC |
| poxB-EcoRV-R | AGCTGGTACCTCGAGTGATTGCCACCCTTTTTACCTTAGC |
| acs(Ec)-F | TTACCTCTTAATTGGAGCTTATGCCACATATTATTAACATCC |
| acs(Ec)-sacI-R | CCACTAGTTCTAGAGAGCTCGCCTACAAACCGTTACCGACTC |
| acs(Ec) *-mF | GTAGTCGAGAAGCCGCTTGAAGAGAAG |
| acs(Ec) *-mR | CTTCTCTTCAAGCGGCTTCTCGACTAC |
| acs(Se)-F | TTACCTCTTAATTGGAGCTTATGCCACATATTATTAACATCCTACAAGGAGAACAAAAGCATGAGCCAAACACATAAACACGC |
| acs(Se)-sacI-R | CCACTAGTTCTAGAGAGCTCGGCATTTATGGTTATGACGG |
| acs(Se) *-mF | GTGGTGGAGAAACCGCTCGAAGAGAAG |
| acs(Se) *-mR | CTTCTCTTCGAGCGGTTTCTCCAC |
| poxB-PJ-F | TGCTGAAAGGAGTGGAATTTACAGCTAGCTCAGTCCTAGGTATTATGCTAGCAACCCTCCGTCAGATGAACTAAAC |
| FWS-mF | GCAGGAAGCAATAACTAGCAT |
| FWS-mR | ATGCTAGTTATTGCTTCCTGCAATAGACCGAGATAGGGTTGAGTG |
| poxB-F112-mF | GCGAAATTGGCAGCGGCTATNNNCAGGAAACCCACC |
| poxB-F112-mR | ATAGCCGCTGCCAATTTCGC |
| poxB-V380-mF | ATGACGCTATTTTCACCTGTGACNNNGGTACGCCAACGGTG |
| poxB-V380-mR | GTCACAGGTGAAAATAGCGTCAT |
| poxB-sg-F | CAAAACACTCGAATCGGCAGGTTTTAGAGCTAGAAATAGC |
| poxB-sg-R | CTGCCGATTCGAGTGTTTTGACTAGTATTATACCTAGGACTGAGC |
| poxB-up-F | TGGGTAGAGCAGGAAGTGAAAGC |
| poxB-up-R | TACAAACCGTTACCGACTCGCACCTGAATGTGATAACGGTAACAAGT |
| poxB-dn-F | TGCGAGTCGGTAACGGTTTGTAGGCGAAAACAAACTGGCTAAGG |
| poxB-dn-R | TATGGGTTGCGGTTGAATACTG |
| pflB-sg-F | GGTGGTATCAAAATGATCGAGTTTTAGAGCTAGAAATAGC |
| pflB-sg-R | TCGATCATTTTGATACCACCACTAGTATTATACCTAGGACTGAGC |
| pflB-up-F | AATGGTCAATGGGGACTAAACG |
| pflB-up-R | GCCTACAAACCGTTACCGACTCGCACCAGGCTGTGGCTAACTTTTCAT |
| pflB-dn-F | TGCGAGTCGGTAACGGTTTGTAGGCTTTCAACTCGCTGACTAAAGAACAG |
| pflB-dn-R | GGTCACCACTTCCTTCATCAAATC |
| tdcE-sg-F | CTGCGTAAAACCCATAACCAGTTTTAGAGCTAGAAATAGC |
| tdcE-sg-R | TGGTTATGGGTTTTACGCAGACTAGTATTATACCTAGGACTGAGC |
| tdcE-up-F | TCTTCGGATTTACGTGTTCTGG |
| tdcE-up-R | GTTGAGGTGTTGACCGCCTTCTCGCCTTCATACGGTGTATAGTT |
| tdcE-dn-F | CGAGAAGGCGGTCAACACCTCAAC |
| tdcE-dn-R | CCCACGGTATGCGAACTG |
| aceE-sg-F | GCACGCAACGAGCAGGATGGGTTTTAGAGCTAGAAATAGC |
| aceE-sg-R | CCATCCTGCTCGTTGCGTGCACTAGTATTATACCTAGGACTGAGC |
| aceE-up-F | TCCAGTATCAGATTGCCGTCAC |
| aceE-up-R | TGACGCAGGTTCACGCTCAACACCTTCTTCAC |
| aceE-dn-F | TGTTGAGCGTGAACCTGCGTCACCACTTCG |
| aceE-dn-R | TTCACCAGGATTTCGGTCACT |
| Designation | Genotype or Description | References |
|---|---|---|
| Plasmids | ||
| pFT24 | A thermal switch vector used in E. coli, p15A, TriR | [35] |
| pFT26 | λcI (ts), PRL::MCS1, PR::MCS2, p15A, TriR | This study |
| pFT26-poxB | λcI (ts), PRL::MCS1, PR::poxB, p15A, TriR | This study |
| pFT26a1B | λcI (ts), PRL::acsEc, PR::poxB, p15A, TriR | This study |
| pFT26a2B | λcI (ts), PRL::acsEc*, PR::poxB, p15A, TriR | This study |
| pFT26a3B | λcI (ts), PRL::acsSe, PR::poxB, p15A, TriR | This study |
| pFT26a4B | λcI (ts), PRL::acsSe*, PR::poxB, p15A, TriR | This study |
| pFT26a1 | λcI (ts), PRL::acsEc, p15A, TriR | This study |
| pFT26a2 | λcI (ts), PRL::acsEc*, p15A, TriR | This study |
| pFT26a3 | λcI (ts), PRL::acsSe, p15A, TriR | This study |
| pFT26a4 | λcI (ts), PRL::acsSe*, p15A, TriR | This study |
| pFTS | A small vector modified from pFT26, p15A, TriR | This study |
| pFTS-poxB | PJ23101::poxB, p15A, TriR | This study |
| pFT26a1B2 | λcI (ts), PRL::acsEc, PR::poxBF112W, p15A, TriR | This study |
| pFT26a1B3 | λcI (ts), PRL::acsEc, PR::poxBF112W, L253F, p15A, TriR | This study |
| pCas | Used for gene knockout in E. coli, KanR | [45] |
| pTargetF | sgRNA, pMB1, SpeR | [45] |
| E. coli strains | ||
| DH5α | Wild-type | Lab stock |
| MG1655 | Wild-type | Lab stock |
| MGF01 | MG1655 ΔpoxBΔpflBΔtdcEΔaceE | This study |
| TWF001 | L-Threonine producing E. coli strain | [46] |
| TWF105 | TWF001 ΔpoxBΔpflBΔldhAΔadhE | [35] |
| TWF115 | TWF105 ΔtdcE | This study |
| TWF115 ΔaceE | TWF105 ΔtdcEΔaceE | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Y.; Zhang, S.; Wang, J.; Yin, L.; Zhang, H.; Wang, Z.; Song, J.; Hu, X.; Wang, X. Metabolic Detoxification of 2-Oxobutyrate by Remodeling Escherichia coli Acetate Bypass. Metabolites 2021, 11, 30. https://doi.org/10.3390/metabo11010030
Fang Y, Zhang S, Wang J, Yin L, Zhang H, Wang Z, Song J, Hu X, Wang X. Metabolic Detoxification of 2-Oxobutyrate by Remodeling Escherichia coli Acetate Bypass. Metabolites. 2021; 11(1):30. https://doi.org/10.3390/metabo11010030
Chicago/Turabian StyleFang, Yu, Shuyan Zhang, Jianli Wang, Lianghong Yin, Hailing Zhang, Zhen Wang, Jie Song, Xiaoqing Hu, and Xiaoyuan Wang. 2021. "Metabolic Detoxification of 2-Oxobutyrate by Remodeling Escherichia coli Acetate Bypass" Metabolites 11, no. 1: 30. https://doi.org/10.3390/metabo11010030
APA StyleFang, Y., Zhang, S., Wang, J., Yin, L., Zhang, H., Wang, Z., Song, J., Hu, X., & Wang, X. (2021). Metabolic Detoxification of 2-Oxobutyrate by Remodeling Escherichia coli Acetate Bypass. Metabolites, 11(1), 30. https://doi.org/10.3390/metabo11010030