
Coupling Mixed Mode Chromatography/ESI Negative MS Detection with Message-Passing Neural Network Modeling for Enhanced Metabolome Coverage and Structural Identification


Abstract
:
1. Introduction
2. Results
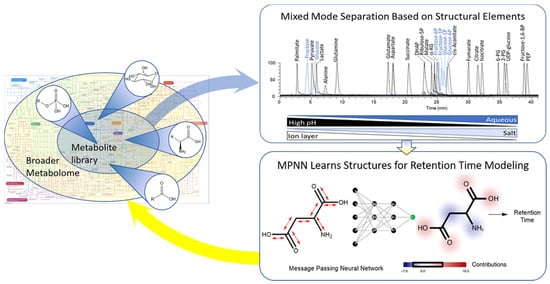
2.1. Mixed Mode Chromatography for Central Carbon Metabolite Detection
2.2. Method Validation Using Purified Metabolites
2.3. Structural Diversity Assessment
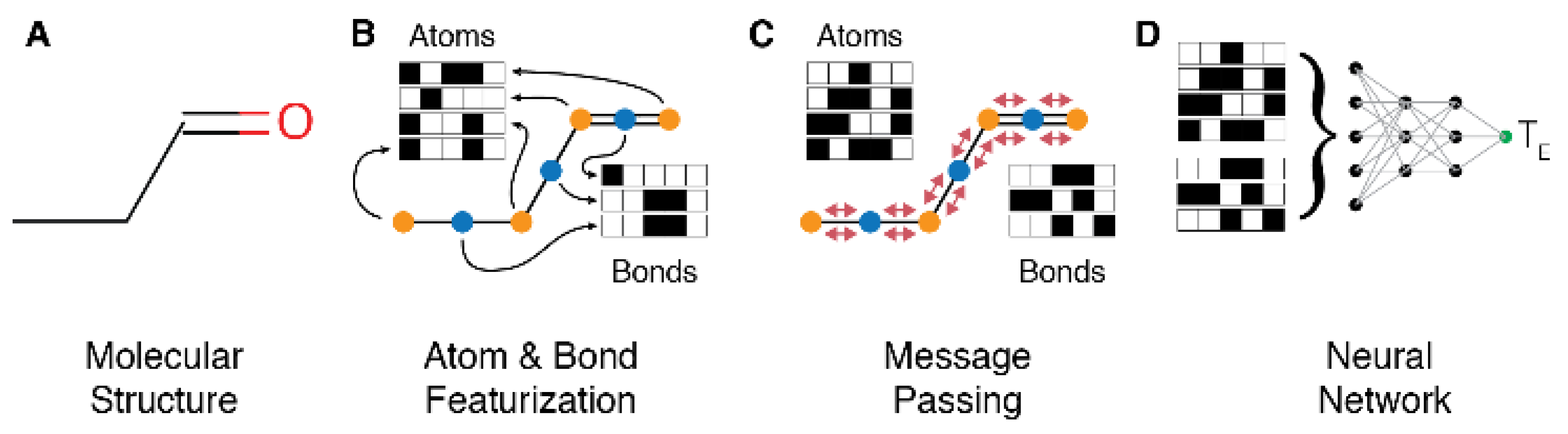
2.4. Mathematical Model for Structure Digitization and Prediction
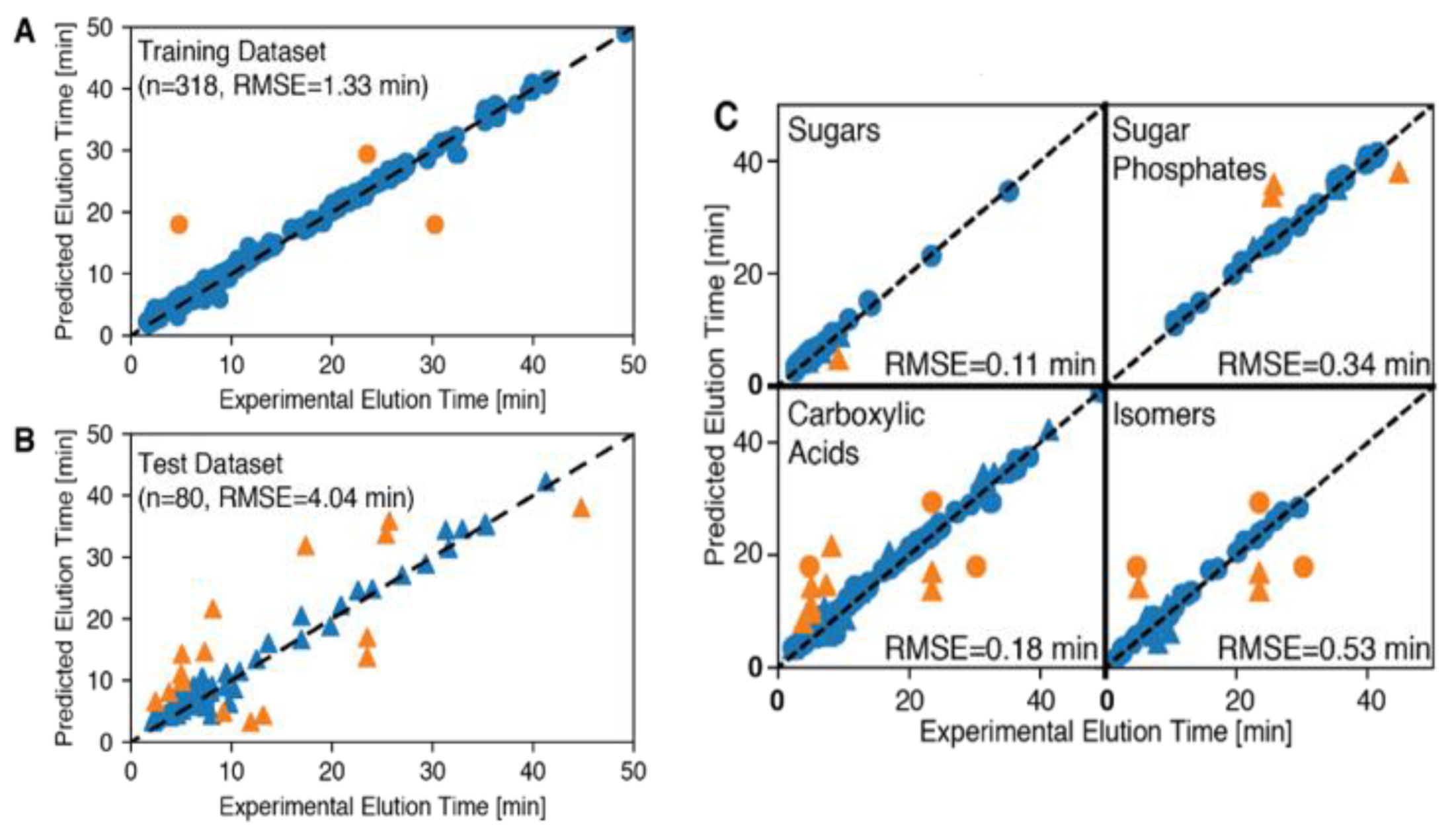
2.4.1. Model Training
2.4.2. Model Performance Predicting RT
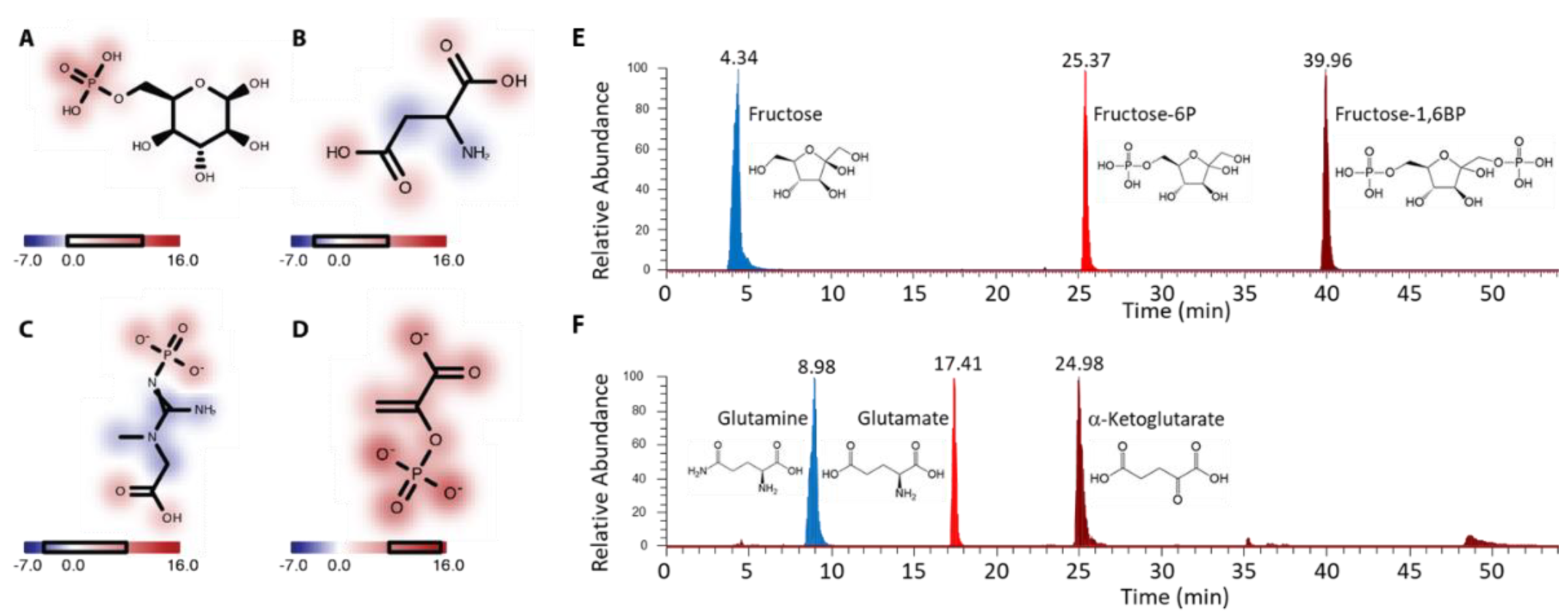
2.4.3. Identification of Important Functional Groups
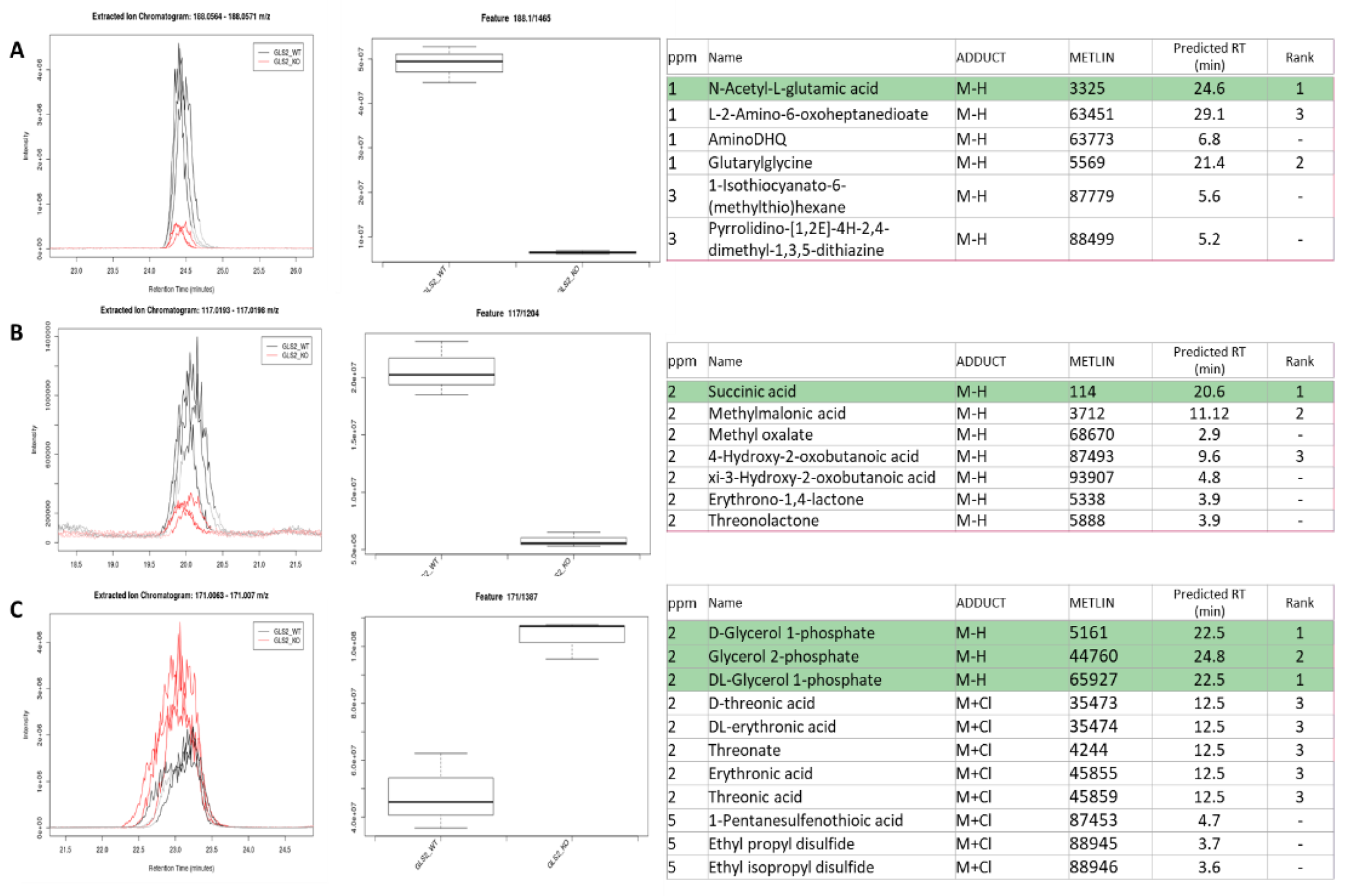
2.5. Application to GLS2 KO Mouse Primary Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Standard Solutions and Sample Preparations
4.3. LCMS
4.4. Hepatocyte and Tissue Isolation and Treatment
4.5. Cell and Tissue Extraction
4.6. Message Passing Neural Network (MPNN)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manier, S.K.; Keller, A.; Schäper, J.; Meyer, M.R. Untargeted metabolomics by high resolution mass spectrometry coupled to normal and reversed phase liquid chromatography as a tool to study the in vitro biotransformation of new psychoactive substances. Sci. Rep. 2019, 9, 2741. [Google Scholar] [CrossRef] [Green Version]
- Schwaiger, M.; Rampler, E.; Hermann, G.; Miklos, W.; Berger, W.; Koellensperger, G. Anion-Exchange Chromatography Coupled to High-Resolution Mass Spectrometry: A Powerful Tool for Merging Targeted and Non-targeted Metabolomics. Anal. Chem. 2017, 89, 7667–7674. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.Q.; Zou, L.; Yin, X.X.; Ong, C.N. HILIC-MS for metabolomics: An attractive and complementary approach to RPLC-MS. Mass Spectrom. Rev. 2016, 35, 574–600. [Google Scholar] [CrossRef]
- Bajad, S.U.; Lu, W.; Kimball, E.H.; Yuan, J.; Peterson, C.; Rabinowitz, J.D. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J. Chromatogr. A 2006, 1125, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Groenke, K.; Takors, R.; Wandrey, C.; Oldiges, M. Simultaneous determination of multiple intracellular metabolites in glycolysis, pentose phosphate pathway and tricarboxylic acid cycle by liquid chromatography-mass spectrometry. J. Chromatogr. A 2007, 1147, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Clasquin, M.F.; Melamud, E.; Amador-Noguez, D.; Caudy, A.A.; Rabinowitz, J.D. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 2010, 82, 3212–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpert, A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. A 1990, 499, 177–196. [Google Scholar] [CrossRef]
- Alpert, A.J. Electrostatic Repulsion Hydrophilic Interaction Chromatography for Isocratic Separation of Charged Solutes and Selective Isolation of Phosphopeptides. Anal. Chem. 2008, 80, 62–76. [Google Scholar] [CrossRef] [Green Version]
- Wamelink, M.M.C.; Struys, E.A.; Huck, J.H.J.; Roos, B.; van der Knaap, M.S.; Jakobs, C.; Verhoeven, N.M. Quantification of sugar phosphate intermediates of the pentose phosphate pathway by LC–MS/MS: Application to two new inherited defects of metabolism. J. Chromatogr. B 2005, 823, 18–25. [Google Scholar] [CrossRef]
- Kiefer, P.; Portais, J.-C.; Vorholt, J.A. Quantitative metabolome analysis using liquid chromatography–high-resolution mass spectrometry. Anal. Biochem. 2008, 382, 94–100. [Google Scholar] [CrossRef]
- Buescher, J.M.; Moco, S.; Sauer, U.; Zamboni, N. Ultrahigh performance liquid chromatography-tandem mass spectrometry method for fast and robust quantification of anionic and aromatic metabolites. Anal. Chem. 2010, 82, 4403–4412. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Tschernutter, V.; Yang, J.; Eckle, T.; Borchers, C.H. Analysis of Selected Sugars and Sugar Phosphates in Mouse Heart Tissue by Reductive Amination and Liquid Chromatography-Electrospray Ionization Mass Spectrometry. Anal. Chem. 2013, 85, 5965–5973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathon, C.; Barding, G.A.; Larive, C.K. Separation of ten phosphorylated mono-and disaccharides using HILIC and ion-pairing interactions. Anal. Chim. Acta 2017, 972 (Suppl. C), 102–110. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huan, T.; Forsberg, E.M.; Rinehart, D.; Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Fang, M.; Aisporna, A.; Hilmers, B.; Poole, F.L.; et al. Systems biology guided by XCMS Online metabolomics. Nat. Methods 2017, 14, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.N.; Belanger, D.; Adams, R.P.; Sculley, D. Rapid Prediction of Electron-Ionization Mass Spectrometry using Neural Networks. ACS Cent. Sci. 2019, 5, 700–708. [Google Scholar] [CrossRef]
- Djoumbou-Feunang, Y.; Pon, A.; Karu, N.; Zheng, J.; Li, C.; Arndt, D.; Gautam, M.; Allen, F.; Wishart, D.S. CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites 2019, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Pirhaji, L.; Milani, P.; Leidl, M.; Curran, T.; Avila-Pacheco, J.; Clish, C.B.; White, F.M.; Saghatelian, A.; Fraenkel, E. Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat. Methods 2016, 13, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Witting, M.; Böcker, S. Current status of retention time prediction in metabolite identification. J. Sep. Sci. 2020, 43, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Fraser, K.; Huege, J.; Featonby, T.; Rasmussen, S.; Jones, C. Predicting retention time in hydrophilic interaction liquid chromatography mass spectrometry and its use for peak annotation in metabolomics. Metab. Off. J. Metab. Soc. 2015, 11, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broeckling, C.D.; Ganna, A.; Layer, M.; Brown, K.; Sutton, B.; Ingelsson, E.; Peers, G.; Prenni, J.E. Enabling Efficient and Confident Annotation of LC-MS Metabolomics Data through MS1 Spectrum and Time Prediction. Anal. Chem. 2016, 88, 9226–9234. [Google Scholar] [CrossRef] [Green Version]
- Randazzo, G.M.; Tonoli, D.; Hambye, S.; Guillarme, D.; Jeanneret, F.; Nurisso, A.; Goracci, L.; Boccard, J.; Rudaz, S. Prediction of retention time in reversed-phase liquid chromatography as a tool for steroid identification. Anal. Chim. Acta 2016, 916, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, A.M.; Lozano, S.; Umbdenstock, T.; Croixmarie, V.; Arrault, A.; Vayer, P. UPLC–MS retention time prediction: A machine learning approach to metabolite identification in untargeted profiling. Metab. Off. J. Metab. Soc. 2015, 12, 8. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Guijas, C.; Billings, E.; Montenegro-Burke, J.R.; Uritboonthai, W.; Aisporna, A.E.; Chen, E.; Benton, H.P.; Siuzdak, G. The METLIN small molecule dataset for machine learning-based retention time prediction. Nat. Commun. 2019, 10, 5811. [Google Scholar] [CrossRef] [PubMed]
- Osipenko, S.; Bashkirova, I.; Sosnin, S.; Kovaleva, O.; Fedorov, M.; Nikolaev, E.; Kostyukevich, Y. Machine learning to predict retention time of small molecules in nano-HPLC. Anal. Bioanal. Chem. 2020, 412, 7767–7776. [Google Scholar] [CrossRef] [PubMed]
- Hetrick, E.M.; Kramer, T.T.; Risley, D.S. Evaluation of a hydrophilic interaction liquid chromatography design space for sugars and sugar alcohols. J. Chromatogr. A 2017, 1489, 65–74. [Google Scholar] [CrossRef]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural Message Passing for Quantum Chemistry. Available online: http://arxiv.org/abs/1704.01212 (accessed on 10 November 2021).
- Vinyals, O.; Bengio, S.; Kudlur, M. Order Matters: Sequence to Sequence for Sets. Available online: http://arxiv.org/abs/1511.06391 (accessed on 10 November 2021).
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef] [Green Version]
- de Koning, L.; Malik, V.S.; Rimm, E.B.; Willett, W.C.; Hu, F.B. Sugar-sweetened and artificially sweetened beverage consumption and risk of type 2 diabetes in men. Am. J. Clin. Nutr. 2011, 93, 1321–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, M.B.; Manson, J.E.; Ludwig, D.S.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. Jama 2004, 292, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Jebb, S.A.; Aveyard, P.; Ambrosini, G.L.; Perez-Cornago, A.; Carter, J.; Sun, X.; Piernas, C. Associations between dietary patterns and the incidence of total and fatal cardiovascular disease and all-cause mortality in 116,806 individuals from the UK Biobank: A prospective cohort study. BMC Med. 2021, 19, 83. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Li, S.; Khan, J.; Dai, Z.; Li, C.; Hu, X.; Shen, Q.; Xue, Y. Sugar- and Artificially Sweetened Beverages Consumption Linked to Type 2 Diabetes, Cardiovascular Diseases, and All-Cause Mortality: A Systematic Review and Dose-Response Meta-Analysis of Prospective Cohort Studies. Nutrients 2021, 13, 2636. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, L.S.; Lacey, J.V., Jr.; Martinez, M.E.; Lemus, H.; Sears, D.D.; Araneta, M.R.G.; Anderson, C.A.M. Association between Sugar-Sweetened Beverage Intake and Mortality Risk in Women: The California Teachers Study. J. Acad. Nutr. Diet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. -Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Sun, X.; Jia, L.; Ma, J.; Xing, H.; Wu, J.; Gao, H.; Sun, Y.; Boulnois, F.; Fan, J. Chemi-Net: A Molecular Graph Convolutional Network for Accurate Drug Property Prediction. Int. J. Mol. Sci. 2019, 20, 3389. [Google Scholar] [CrossRef] [Green Version]
- Melamud, E.; Vastag, L.; Rabinowitz, J.D. Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 2010, 82, 9818–9826. [Google Scholar] [CrossRef] [Green Version]
- Clasquin, M.F.; Melamud, E.; Rabinowitz, J.D. LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinform. 2012, 37. [Google Scholar]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [Green Version]
- Deepchem API. Available online: https://deepchem.readthedocs.io/en/latest/api_reference/models.html#mpnnmodel (accessed on 10 November 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | LOD (pmole) | CV (n = 4) | Linear Range (pmole) | Linear Coefficient (R2) | Mass Error (ppm) |
|---|---|---|---|---|---|
| 2-phosphoglycerate | 0.0254 | 10.8 | 0.229–1000 | 0.98729 | 0.54 |
| 6-phosphogluconate | 0.076 | 10.7 | 0.229–500 | 0.9993 | 0 |
| a-ketoglutarate | 0.685 | 15.8 | 2.06–1500 | 0.99622 | 0.69 |
| L-alanine | 6.15 | 20.9 | 6.15–1500 | 0.99199 | 2.27 |
| L-aspartate | 0.076 | 9.15 | 0.229–1500 | 0.99862 | 0 |
| cis-aconitate | 0.0254 | 39.3 | 0.0760–166.5 | 0.99015 | 0.58 |
| DHAP | 18.5 | 33.2 | 18.5–1500 | 0.99815 | 0.59 |
| sedoheptulose 7P | 0.076 | 12.8 | 0.229–500 | 0.9981 | 0.69 |
| fructose-13C6 | 0.0254 | 12 | 0.0254–166.5 | 0.99749 | 0.54 |
| fructose-1,6-BP | 0.0254 | 17.6 | 0.0254–1500 | 0.99259 | 0.59 |
| fructose-1P | 0.0254 | 9.62 | 0.0760–1500 | 0.99408 | 0 |
| fructose-6P | 0.0254 | 12.8 | 0.0760–1500 | 0.99078 | 0 |
| fumarate | 0.685 | 11.3 | 2.06–1500 | 0.98993 | 0 |
| glucose | 0.685 | 13.8 | 2.06–1500 | 0.98751 | 0.56 |
| glucose-1P | 0.0254 | 12.3 | 0.0760–500 | 0.9955 | 0 |
| glucose-6P | 0.0254 | 11.4 | 0.0760–500 | 0.99585 | 0 |
| glutamate | 0.0254 | 8.95 | 0.229–3000 | 0.99572 | −0.68 |
| glutamine | 0.076 | 6.85 | 0.229–1500 | 0.99001 | −0.69 |
| glyceraldehyde-3P | 55.5 | 5.45 | 55.5–1500 | 0.98939 | 0.59 |
| glycine | 18.5 | 4.71 | 18.5–1500 | 0.99478 | 0 |
| glyecrol-3P | 0.076 | 7.95 | 0.229–500 | 0.99723 | 0.58 |
| isocitrate | 6.15 | 31.6 | 6.15–500 | 0.96498 | 0.52 |
| lactate | 0.229 | z12.4 | 0.229–55.5 | 0.98791 | 0 |
| malate | 0.076 | 39.2 | 0.229–166.5 | 0.99544 | 0.75 |
| myo-inositol-d6 | 6.15 | 15.1 | 6.15–1500 | 0.9838 | 0.54 |
| phosphoenolpyruvate | 0.076 | 14.3 | 0.229–1500 | 0.99564 | 0.6 |
| pyruvate | 2.06 | 18.9 | 2.06–1000 | 0.96721 | 0 |
| ribulose-5P | 0.685 | 9.84 | 0.685–1000 | 0.98069 | −0.44 |
| serine | 0.229 | 9.88 | 0.229–1500 | 0.99155 | 0.96 |
| sorbitol | 0.076 | 11.2 | 0.229–500 | 0.99293 | 0 |
| succinate | 0.229 | 12.2 | 2.06–1000 | 0.96498 | 0 |
| UDP-glucose | 0.229 | 12.1 | 0.229–1500 | 0.99135 | 1.77 |
| xylitol-13C5 | 0.229 | 9.35 | 2.06–1000 | 0.98754 | 0.64 |
| Training | Test | |||
|---|---|---|---|---|
| Subset | Dataset Size | # Points with Error <2 min | Dataset Size | # Points with Error <2 min |
| Sugars | 55 | 55 | 11 | 10 |
| Sugar-P | 39 | 39 | 6 | 3 |
| Carboxylic acids | 148 | 145 | 35 | 27 |
| Isomers | 37 | 34 | 11 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, G.; Sresht, V.; Sun, Z.; Shi, Y.; Clasquin, M.F. Coupling Mixed Mode Chromatography/ESI Negative MS Detection with Message-Passing Neural Network Modeling for Enhanced Metabolome Coverage and Structural Identification. Metabolites 2021, 11, 772. https://doi.org/10.3390/metabo11110772
Xing G, Sresht V, Sun Z, Shi Y, Clasquin MF. Coupling Mixed Mode Chromatography/ESI Negative MS Detection with Message-Passing Neural Network Modeling for Enhanced Metabolome Coverage and Structural Identification. Metabolites. 2021; 11(11):772. https://doi.org/10.3390/metabo11110772
Chicago/Turabian StyleXing, Gang, Vishnu Sresht, Zhongyuan Sun, Yuji Shi, and Michelle F. Clasquin. 2021. "Coupling Mixed Mode Chromatography/ESI Negative MS Detection with Message-Passing Neural Network Modeling for Enhanced Metabolome Coverage and Structural Identification" Metabolites 11, no. 11: 772. https://doi.org/10.3390/metabo11110772