
A Core Metabolome Response of Maize Leaves Subjected to Long-Duration Abiotic Stresses

 , , , and
, , , and 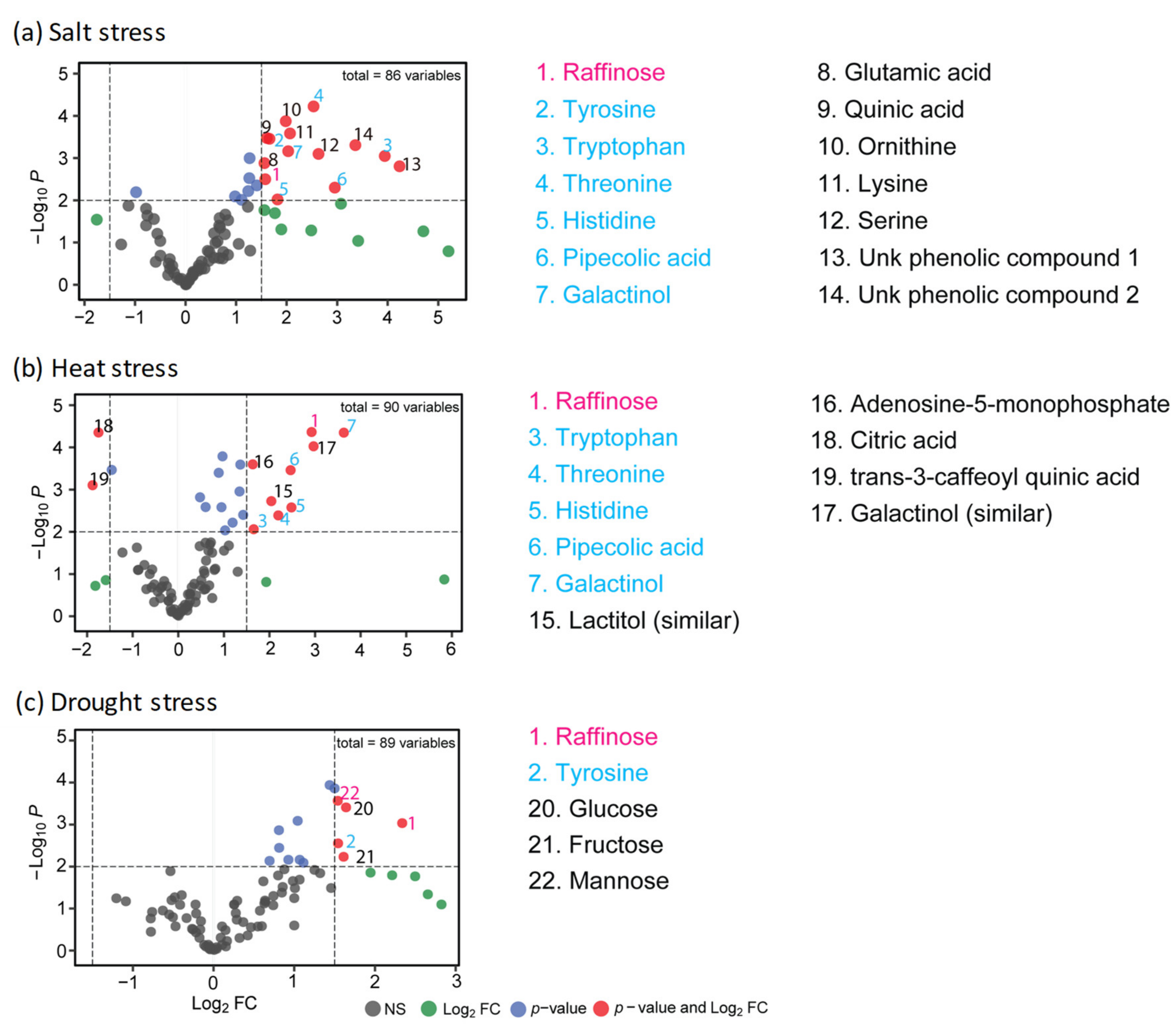{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
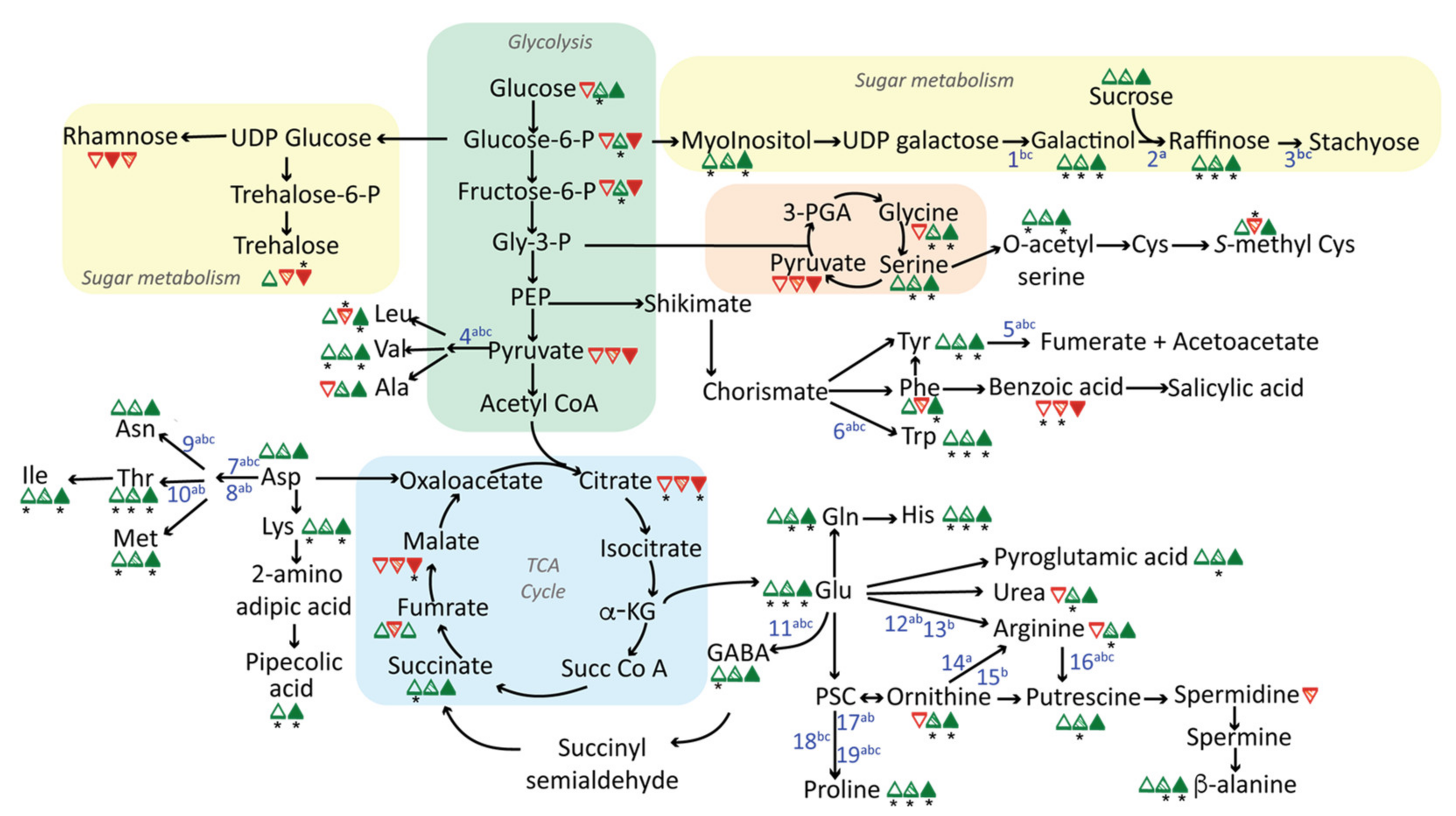
2.1. Changes in Central Metabolism in Response to Long-Term Drought, Heat, or Salt Stress
2.2. Shared Features of Stress Metabolomes
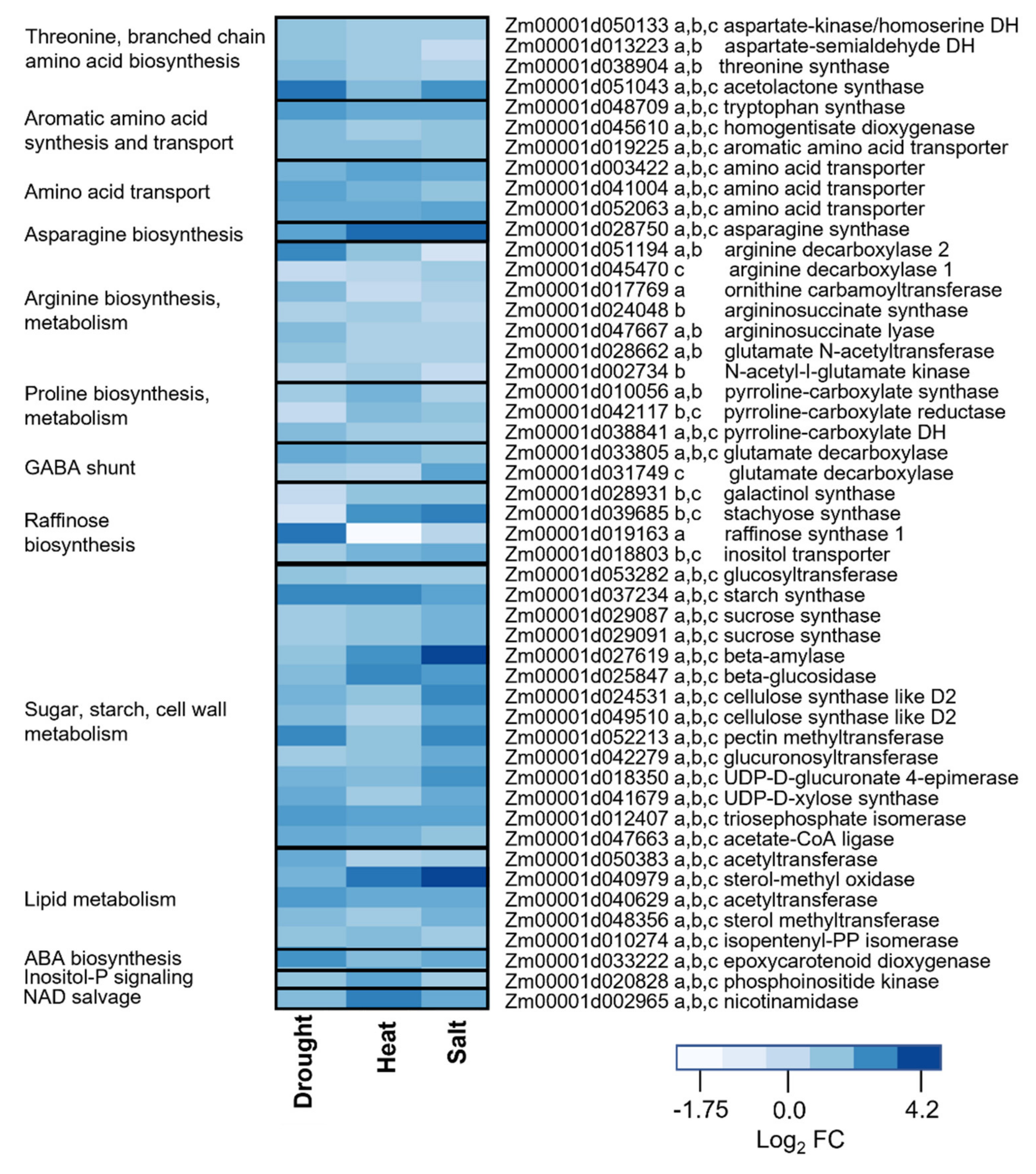
2.3. Transcriptome Changes Underlying of Metabolic Reprogramming of Stressed Leaves
3. Discussion
3.1. Prominent Roles of Amino Acids in Stress Metabolomes
3.2. GABA Shunt Activation as a Thiamin-Deficiency Workaround?
3.3. Upregulation of the Raffinose Biosynthetic Pathway
3.4. Multiple Signaling Mechanisms Contribute to Shared Metabolic Responses to Stress
4. Materials and Methods
4.1. Plant Growth Conditions
4.2. Drought Stress
4.3. Heat Stress
4.4. Salt Stress
4.5. Metabolite Analysis
4.6. RNAseq
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, R.; Bayer, P.E.; Edwards, D. Climate change and the need for agricultural adaptation. Curr. Opin. Plant Biol. 2020, 56, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Hanson, A.D.; Hitz, W.D. Metabolic responses of mesophytes to plant water deficits. Annu. Rev. Plant Physiol. 1982, 33, 163–203. [Google Scholar] [CrossRef]
- Crafts-Brandner, S.J.; Salvucci, M.E. Sensitivity of photosynthesis in a C4 plant, maize, to heat stress. Plant Physiol. 2002, 129, 1773–1780. [Google Scholar] [CrossRef] [Green Version]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [Green Version]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef] [Green Version]
- Hussain, H.A.; Men, S.; Hussain, S.; Chen, Y.; Ali, S.; Zhang, S.; Zhang, K.; Li, Y.; Xu, Q.; Liao, C.; et al. Interactive effects of drought and heat stresses on morpho-physiological attributes, yield, nutrient uptake and oxidative status in maize hybrids. Sci. Rep. 2019, 9, 3890. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Li, X.; Niu, L.; Jameson, P.E.; Zhou, W. Transcription-associated metabolomic adjustments in maize occur during combined drought and cold stress. Plant Physiol. 2021, 186, 677–695. [Google Scholar] [CrossRef]
- Szabados, L.; Savoure, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Székely, G.; Abrahám, E.; Cséplo, A.; Rigó, G.; Zsigmond, L.; Csiszár, J.; Ayaydin, F.; Strizhov, N.; Jásik, J.; Schmelzer, E.; et al. Duplicated P5CS genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J. 2008, 53, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.; Honig, A.; Stein, H.; Suzuki, N.; Mittler, R.; Zilberstein, A. Unraveling Δ1-pyrroline-5-carboxylate-proline cycle in plants by uncoupled expression of proline oxidation enzymes. J. Biol. Chem. 2009, 284, 26482–26492. [Google Scholar] [CrossRef] [Green Version]
- Renault, H.; Roussel, V.; El Amrani, A.; Arzel, M.; Renault, D.; Bouchereau, A.; Deleu, C. The Arabidopsis pop2-1 mutant reveals the involvement of GABA transaminase in salt stress tolerance. BMC Plant Biol. 2010, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Michaeli, S.; Fromm, H. Closing the loop on the GABA shunt in plants: Are GABA metabolism and signaling entwined? Front. Plant Sci. 2015, 6, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, T.B.; Chapman, L.M. The importance of thiamine (vitamin B1) in plant health: From crop yield to biofortification. J. Biol. Chem. 2020, 295, 12002–12013. [Google Scholar] [CrossRef]
- Hanson, A.D.; Beaudoin, G.A.; McCarty, D.R.; Gregory, J.F., 3rd. Does abiotic stress cause functional B vitamin deficiency in plants? Plant Physiol. 2016, 172, 2082–2097. [Google Scholar] [CrossRef] [Green Version]
- Joshi, J.; Folz, J.S.; Gregory, J.F., 3rd; McCarty, D.R.; Fiehn, O.; Hanson, A.D. Rethinking the PDH bypass and GABA shunt as thiamin-deficiency workarounds. Plant Physiol. 2019, 181, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Mundree, S.G.; Thomson, J.A.; Farrant, J.M.; Keller, F. Protection mechanisms in the resurrection plant Xerophyta viscosa (Baker): Both sucrose and raffinose family oligosaccharides (RFOs) accumulate in leaves in response to water deficit. J. Exp. Bot. 2007, 58, 1947–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizawa, A.; Yabuta, Y.; Shigeoka, S. Galactinol and raffinose constitute a novel function to protect plants from oxidative damage. Plant Physiol. 2008, 147, 1251–1263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, Q.; Zhang, C.; Hao, G.; Wang, C.; Dirk, L.M.; Downie, A.B.; Zhao, T. Maize VIVIPAROUS1 interacts with ABA INSENSITIVE5 to regulate GALACTINOL SYNTHASE2 expression controlling seed raffinose accumulation. J. Agric. Food Chem. 2019, 67, 4214–4223. [Google Scholar] [CrossRef]
- Yang, S.; Vanderbeld, B.; Wan, J.; Huang, Y. Narrowing down the targets: Towards successful genetic engineering of drought-tolerant crops. Mol. Plant 2010, 3, 469–490. [Google Scholar] [CrossRef]
- Blum, A. Genomics for drought resistance—Getting down to earth. Funct. Plant Biol. 2014, 41, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Makarevitch, I.; Waters, A.J.; West, P.T.; Stitzer, M.; Hirsch, C.N.; Ross-Ibarra, J.; Springer, N.M. Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 2015, 11, e1004915. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Rangel, M.R.; Chávez Montes, R.A.; González-Segovia, E.; Ross-Ibarra, J.; Simpson, J.K.; Sawers, R.J.H. Allele specific expression analysis identifies regulatory variation associated with stress-related genes in the Mexican highland maize landrace Palomero Toluqueño. PeerJ 2017, 5, e3737. [Google Scholar] [CrossRef] [Green Version]
- Fàbregas, N.; Fernie, A.R. The metabolic response to drought. J. Exp. Bot. 2019, 70, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villate, A.; San Nicolas, M.; Gallastegi, M.; Aulas, P.A.; Olivares, M.; Usobiaga, A.; Etxebarria, N.; Aizpurua-Olaizola, O. Review: Metabolomics as a prediction tool for plants performance under environmental stress. Plant Sci. 2021, 303, 110789. [Google Scholar] [CrossRef]
- Li, P.; Cao, W.; Fang, H.; Xu, S.; Yin, S.; Zhang, Y.; Lin, D.; Wang, J.; Chen, Y.; Xu, C.; et al. Transcriptomic profiling of the maize (Zea mays L.) leaf response to abiotic stresses at the seedling stage. Front. Plant Sci. 2017, 8, 290. [Google Scholar] [CrossRef] [Green Version]
- Hackett, S.R.; Zanotelli, V.R.; Xu, W.; Goya, J.; Park, J.O.; Perlman, D.H.; Gibney, P.A.; Botstein, D.; Storey, J.D.; Rabinowitz, J.D. Systems-level analysis of mechanisms regulating yeast metabolic flux. Science 2016, 354, aaf2786. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, D.; Sun, J.; Zhang, A. Asparagine synthetase gene TaASN1 from wheat is up-regulated by salt stress, osmotic stress and ABA. J. Plant Physiol. 2005, 162, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Zhang, F.; Xu, Y.; Chen, X.; Qin, X.; Wen, X. Effect of post-silking drought on nitrogen partitioning and gene expression patterns of glutamine synthetase and asparagine synthetase in two maize (Zea mays L.) varieties. Plant Physiol. Biochem. 2016, 102, 62–69. [Google Scholar] [CrossRef]
- Xu, B.; Long, Y.; Feng, X.; Zhu, X.; Sai, N.; Chirkova, L.; Betts, A.; Herrmann, J.; Edwards, E.J.; Okamoto, M.; et al. GABA signalling modulates stomatal opening to enhance plant water use efficiency and drought resilience. Nat. Commun. 2021, 12, 1952. [Google Scholar] [CrossRef]
- Zhuo, C.; Wang, T.; Lu, S.; Zhao, Y.; Li, X.; Guo, Z. A cold responsive galactinol synthase gene from Medicago falcata (MfGolS1) is induced by myo-inositol and confers multiple tolerances to abiotic stresses. Physiol. Plant 2013, 149, 67–78. [Google Scholar] [CrossRef]
- Maas, E.V.; Grieve, C.M. Sodium-induced calcium deficiency in salt-stressed corn. Plant Cell Environ. 1987, 10, 559–564. [Google Scholar] [CrossRef]
- Grieve, C.M.; Grattan, S.R.; Maas, E.V. Plant salt tolerance. In ASCE Manual and Reports on Engineering Practice No. 71 Agricultural Salinity Assessment and Management, 2nd ed.; Wallender, W.W., Tanji, K.K., Eds.; ASCE: Reston, VA, USA, 2012; pp. 405–459. [Google Scholar]
- Suzuki, M.; Wu, S.; Mimura, M.; Alseekh, S.; Fernie, A.R.; Hanson, A.D.; McCarty, D.R. Construction and applications of a B vitamin genetic resource for investigation of vitamin-dependent metabolism in maize. Plant J. 2020, 101, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Roessner, U.; Luedemann, A.; Brust, D.; Fiehn, O.; Linke, T.; Willmitzer, L.; Fernie, A. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 2001, 13, 11–29. [Google Scholar] [CrossRef] [Green Version]
- Schauer, N.; Steinhauser, D.; Strelkov, S.; Schomburg, D.; Allison, G.; Moritz, T.; Lundgren, K.; Roessner-Tunali, U.; Forbes, M.G.; Willmitzer, L.; et al. GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett. 2005, 579, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Mimura, M.; Zallot, R.; Niehaus, T.D.; Hasnain, G.; Gidda, S.K.; Nguyen, T.N.; Anderson, E.M.; Mullen, R.T.; Brown, G.; Yakunin, A.F.; et al. Arabidopsis TH2 encodes the orphan enzyme thiamin monophosphate phosphatase. Plant Cell 2016, 28, 2683–2696. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentael, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, J.; Hasnain, G.; Logue, T.; Lynch, M.; Wu, S.; Guan, J.-C.; Alseekh, S.; Fernie, A.R.; Hanson, A.D.; McCarty, D.R. A Core Metabolome Response of Maize Leaves Subjected to Long-Duration Abiotic Stresses. Metabolites 2021, 11, 797. https://doi.org/10.3390/metabo11110797
Joshi J, Hasnain G, Logue T, Lynch M, Wu S, Guan J-C, Alseekh S, Fernie AR, Hanson AD, McCarty DR. A Core Metabolome Response of Maize Leaves Subjected to Long-Duration Abiotic Stresses. Metabolites. 2021; 11(11):797. https://doi.org/10.3390/metabo11110797
Chicago/Turabian StyleJoshi, Jaya, Ghulam Hasnain, Taylor Logue, Madeline Lynch, Shan Wu, Jiahn-Chou Guan, Saleh Alseekh, Alisdair R. Fernie, Andrew D. Hanson, and Donald R. McCarty. 2021. "A Core Metabolome Response of Maize Leaves Subjected to Long-Duration Abiotic Stresses" Metabolites 11, no. 11: 797. https://doi.org/10.3390/metabo11110797