
New Insights to the Crosstalk between Vascular and Bone Tissue in Chronic Kidney Disease–Mineral and Bone Disorder

Abstract
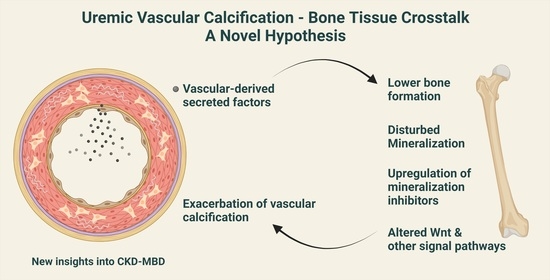
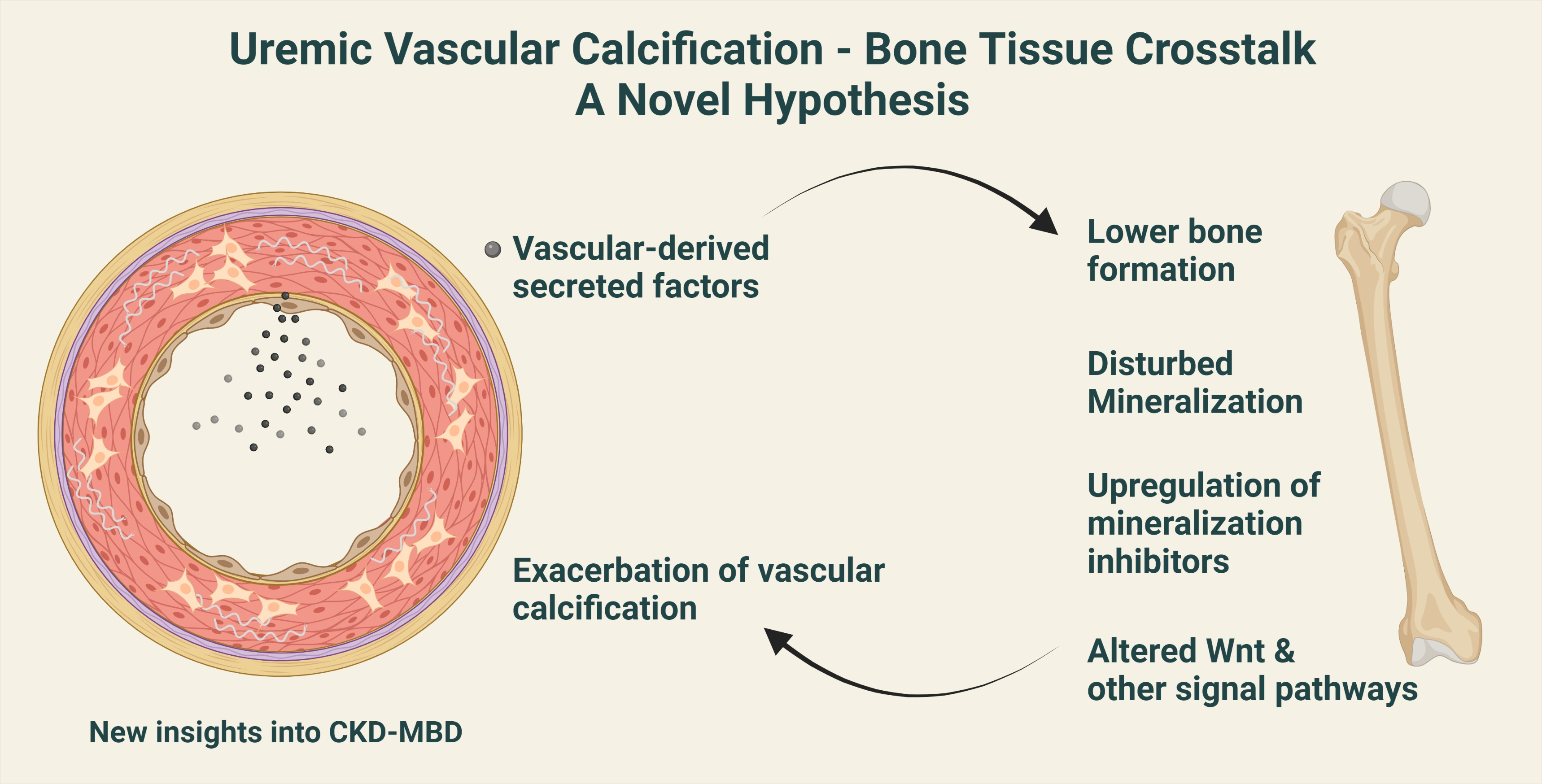
1. Introduction
2. Complex Paracrine Interplay between Bone Vasculature Cells and Bone Cells in the Development and Maintenance of Bone Tissue
3. Dramatic Changes in the Vasculature in CKD
4. Endothelial Dysfunction and Development of Atherosclerosis in CKD
5. Endothelial Dysfunction in CKD Vascular Calcification—An Aspect of Osteomimicry?
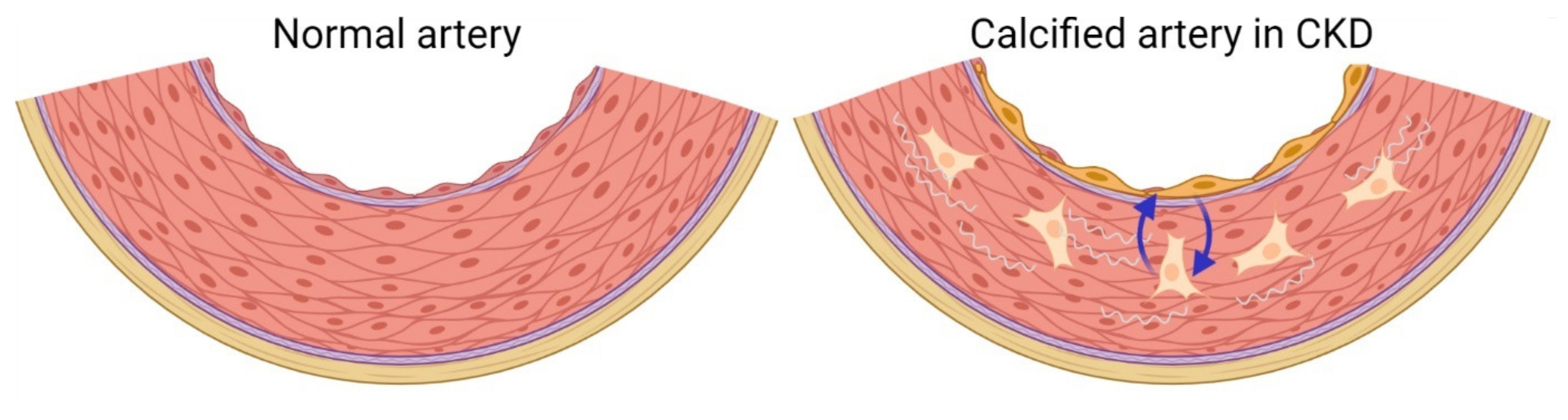
6. Development of Vascular Medial Calcification in CKD
7. New Concept of the Biological Systems Pathology in CKD-MBD
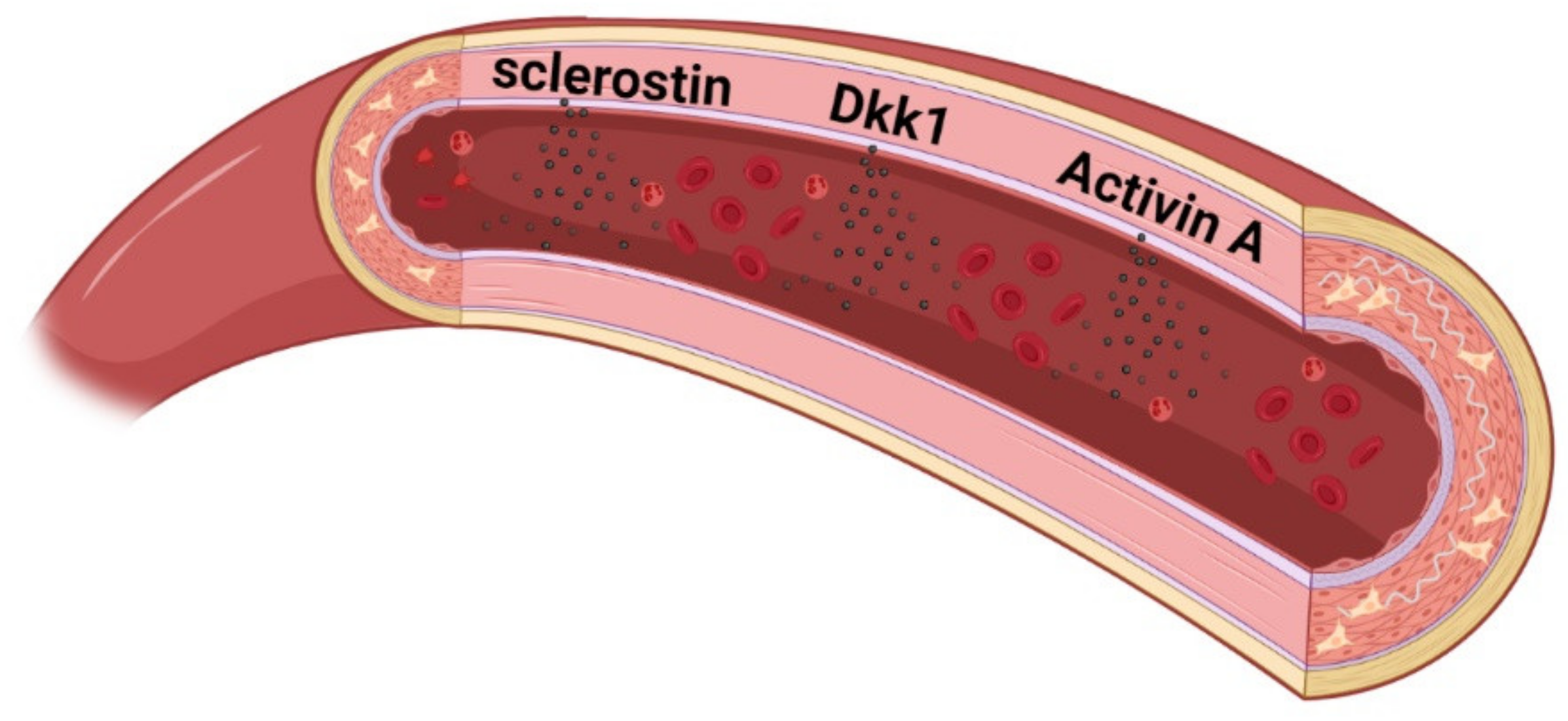
8. New Factors Identified to Play a Role in Vascular Disease in CKD
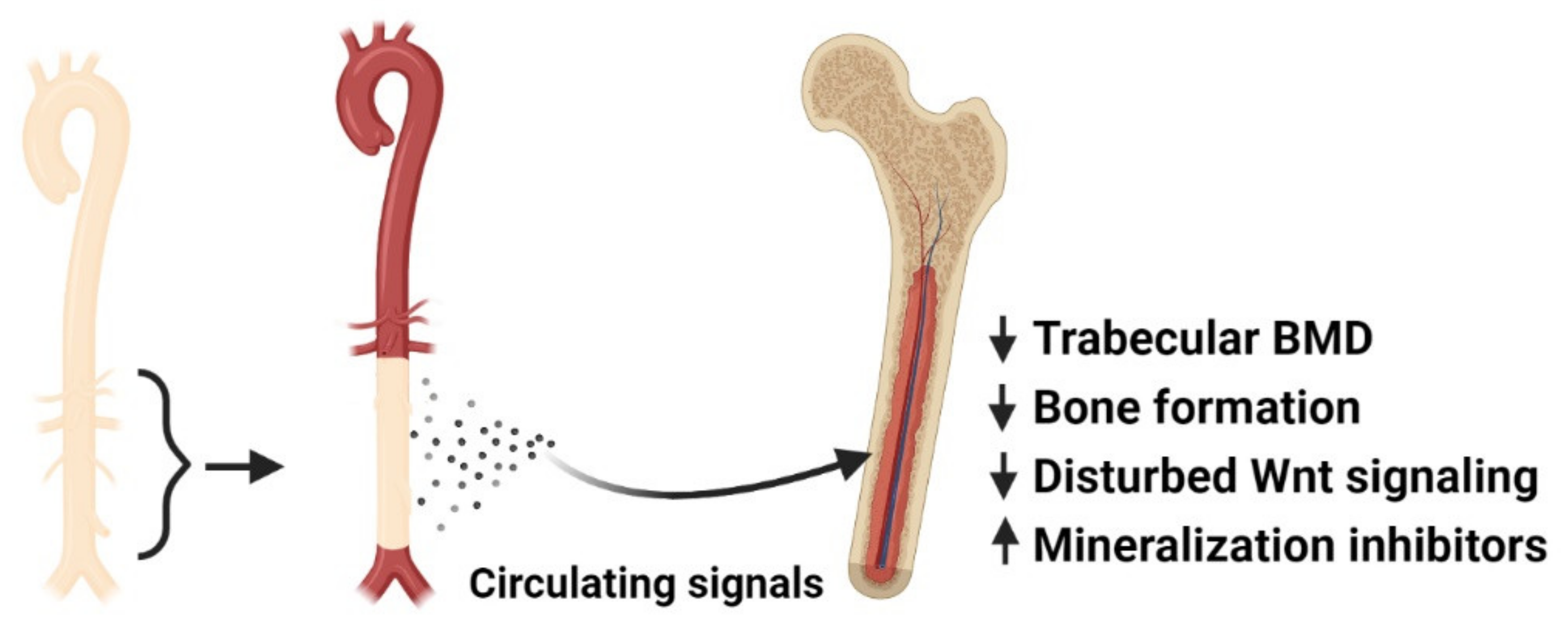
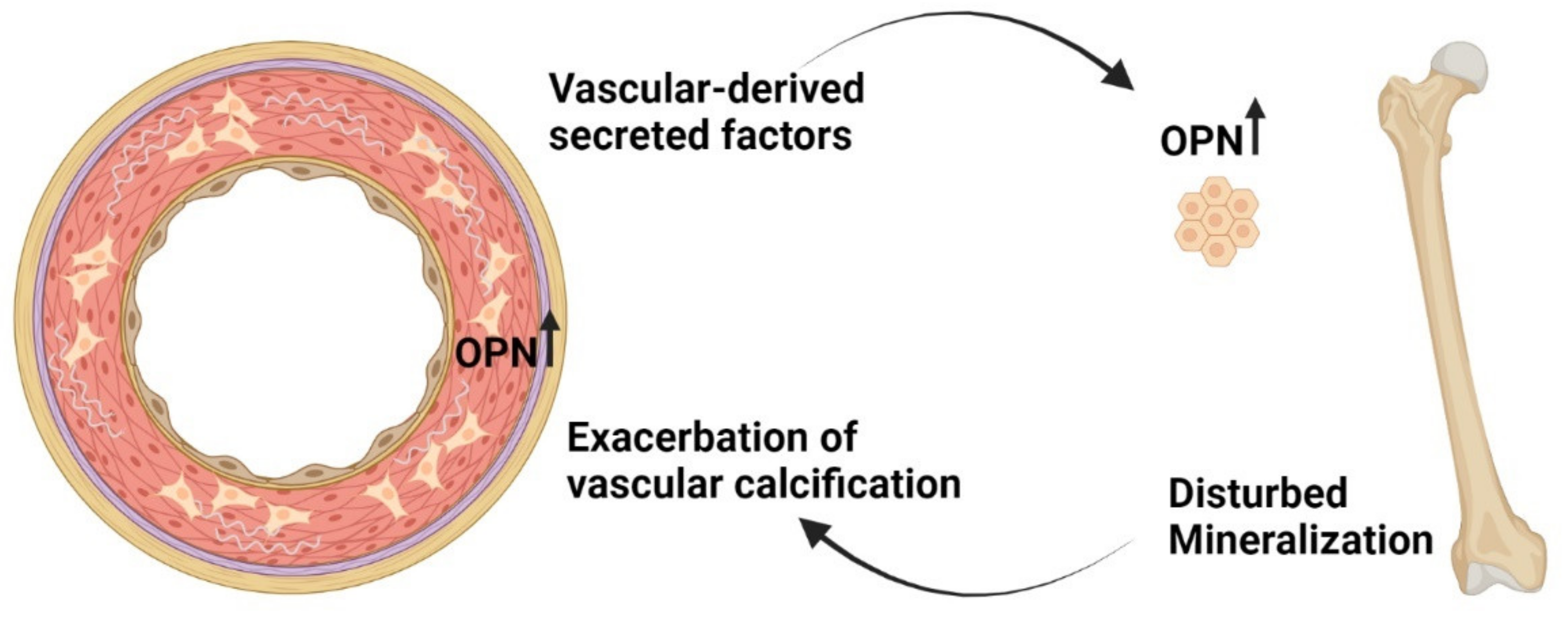
9. Calcified Vasculature Affects Bone Metabolism
10. Can the ‘Calcification Paradox’ Be Explained by the Vascular–Bone Tissue Crosstalk?
11. Disturbances in Wnt Pathway in Renal Osteodystrophy
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed]
- Robling, A.G.; Bonewald, L.F. The Osteocyte: New Insights. Annu. Rev. Physiol. 2020, 82, 485–506. [Google Scholar] [CrossRef] [PubMed]
- Kiran, S.; Kumar, V.; Kumar, S.; Price, R.L.; Singh, U.P. Adipocyte, Immune Cells, and miRNA Crosstalk: A Novel Regulator of Metabolic Dysfunction and Obesity. Cells 2021, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Peng, G.; Zhang, N.; Wang, B.; Luo, B. Crosstalk Between the Gut Microbiota and the Brain: An Update on Neuroimaging Findings. Front. Neurol. 2019, 10, 883. [Google Scholar] [CrossRef]
- Romero, A.; Eckel, J. Organ Crosstalk and the Modulation of Insulin Signaling. Cells 2021, 10, 2082. [Google Scholar] [CrossRef]
- Rehman, A.; Pacher, P.; Hasko, G. Role of Macrophages in the Endocrine System. Trends Endocrinol. Metab. 2021, 32, 238–256. [Google Scholar] [CrossRef]
- Wang, Z.; Pu, Q.; Huang, C.; Wu, M. Crosstalk Between Lung and Extrapulmonary Organs in Infection and Inflammation. Adv. Exp. Med. Biol. 2021, 1303, 333–350. [Google Scholar]
- Jahng, J.W.; Song, E.; Sweeney, G. Crosstalk between the heart and peripheral organs in heart failure. Exp. Mol. Med. 2016, 48, e217. [Google Scholar] [CrossRef]
- Sivaraj, K.K.; Adams, R.H. Blood vessel formation and function in bone. Development 2016, 143, 2706–2715. [Google Scholar] [CrossRef]
- Hyder, J.A.; Allison, M.A.; Criqui, M.H.; Wright, C.M. Association between systemic calcified atherosclerosis and bone density. Calcif. Tissue Int. 2007, 80, 301–306. [Google Scholar] [CrossRef]
- Edmonds, M.E. Medial arterial calcification and diabetes mellitus. Z. Kardiol. 2000, 89 (Suppl. 2), 101–104. [Google Scholar] [CrossRef]
- Hak, A.E.; Pols, H.A.; van Hemert, A.M.; Hofman, A.; Witteman, J.C. Progression of aortic calcification is associated with metacarpal bone loss during menopause: A population-based longitudinal study. Arter. Thromb. Vasc. Biol. 2000, 20, 1926–1931. [Google Scholar] [CrossRef]
- Laroche, M.; Delmotte, A. Increased arterial calcification in Paget’s disease of bone. Calcif. Tissue Int. 2005, 77, 129–133. [Google Scholar] [CrossRef]
- Toussaint, N.D.; Lau, K.K.; Strauss, B.J.; Polkinghorne, K.R.; Kerr, P.G. Associations between vascular calcification, arterial stiffness and bone mineral density in chronic kidney disease. Nephrol. Dial. Transplant. 2008, 23, 586–593. [Google Scholar] [CrossRef]
- Boukhris, R.; Becker, K.L. Calcification of the aorta and osteoporosis. A roentgenographic study. JAMA 1972, 219, 1307–1311. [Google Scholar] [CrossRef]
- Frye, M.A.; Melton, L.J., III; Bryant, S.C.; Fitzpatrick, L.A.; Wahner, H.W.; Schwartz, R.S.; Riggs, B.L. Osteoporosis and calcification of the aorta. Bone Miner. 1992, 19, 185–194. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Moe, S.; Drueke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, H.S.; David, K.; Salam, S.; Evenepoel, P.; European Renal Osteodystrophy Workgroup; Initiative of ERA CKD-MBD Working Group. Traditional and Non-traditional Risk Factors for Osteoporosis in CKD. Calcif. Tissue Int. 2021, 108, 496–511. [Google Scholar] [CrossRef]
- Stegen, S.; Carmeliet, G. The skeletal vascular system—Breathing life into bone tissue. Bone 2018, 115, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, Y.; Huang, X.; Gu, Y.; Li, S.; Yin, P.; Zhang, L.; Tang, P. Skeleton-vasculature chain reaction: A novel insight into the mystery of homeostasis. Bone Res. 2021, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Riancho, J.A.; Salas, E.; Zarrabeitia, M.T.; Olmos, J.M.; Amado, J.A.; Fernandez-Luna, J.L.; Gonzalez-Macias, J. Expression and functional role of nitric oxide synthase in osteoblast-like cells. J. Bone Miner. Res. 1995, 10, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, M.H.; Evans, D.E.; Grabowski, P.S.; Pollock, J.S.; Ohshima, H.; Ralston, S.H. Expression of nitric oxide synthase isoforms in bone and bone cell cultures. J. Bone Miner. Res. 1997, 12, 1108–1115. [Google Scholar] [CrossRef]
- Lin, I.C.; Smartt, J.M., Jr.; Nah, H.D.; Ischiropoulos, H.; Kirschner, R.E. Nitric oxide stimulates proliferation and differentiation of fetal calvarial osteoblasts and dural cells. Plast. Reconstr. Surg. 2008, 121, 1554–1566. [Google Scholar] [CrossRef]
- Veeriah, V.; Zanniti, A.; Paone, R.; Chatterjee, S.; Rucci, N.; Teti, A.; Capulli, M. Interleukin-1beta, lipocalin 2 and nitric oxide synthase 2 are mechano-responsive mediators of mouse and human endothelial cell-osteoblast crosstalk. Sci Rep. 2016, 6, 29880. [Google Scholar] [CrossRef]
- Kalyanaraman, H.; Schall, N.; Pilz, R.B. Nitric oxide and cyclic GMP functions in bone. Nitric Oxide 2018, 76, 62–70. [Google Scholar] [CrossRef]
- Clarkin, C.E.; Emery, R.J.; Pitsillides, A.A.; Wheeler-Jones, C.P. Evaluation of VEGF-mediated signaling in primary human cells reveals a paracrine action for VEGF in osteoblast-mediated crosstalk to endothelial cells. J. Cell. Physiol. 2008, 214, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, K.A.; Raisz, L.G.; Pilbeam, C.C. Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol. Metab. 2010, 21, 294–301. [Google Scholar] [CrossRef] [PubMed]
- von Schroeder, H.P.; Veillette, C.J.; Payandeh, J.; Qureshi, A.; Heersche, J.N. Endothelin-1 promotes osteoprogenitor proliferation and differentiation in fetal rat calvarial cell cultures. Bone 2003, 33, 673–684. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; Yasuda, H.; Yano, K.; Morinaga, T.; Higashio, K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1998, 253, 395–400. [Google Scholar] [CrossRef]
- Collin-Osdoby, P.; Rothe, L.; Anderson, F.; Nelson, M.; Maloney, W.; Osdoby, P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J. Biol. Chem. 2001, 276, 20659–20672. [Google Scholar] [CrossRef]
- Rosen, V. BMP2 signaling in bone development and repair. Cytokine Growth Factor Rev. 2009, 20, 475–480. [Google Scholar] [CrossRef]
- Bouletreau, P.J.; Warren, S.M.; Spector, J.A.; Peled, Z.M.; Gerrets, R.P.; Greenwald, J.A.; Longaker, M.T. Hypoxia and VEGF up-regulate BMP-2 mRNA and protein expression in microvascular endothelial cells: Implications for fracture healing. Plast. Reconstr. Surg. 2002, 109, 2384–2397. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Shang, X.; Zhang, H.; Wang, G.; Massey, P.A.; Barton, S.R.; Kevil, C.G.; Dong, Y. Notch Signaling in Osteogenesis, Osteoclastogenesis, and Angiogenesis. Am. J. Pathol. 2019, 189, 1495–1500. [Google Scholar] [CrossRef]
- Yang, X.; Tare, R.S.; Partridge, K.A.; Roach, H.I.; Clarke, N.M.; Howdle, S.M.; Shakesheff, K.M.; Oreffo, R.O. Induction of human osteoprogenitor chemotaxis, proliferation, differentiation, and bone formation by osteoblast stimulating factor-1/pleiotrophin: Osteoconductive biomimetic scaffolds for tissue engineering. J. Bone Miner. Res. 2003, 18, 47–57. [Google Scholar] [CrossRef]
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Karsenty, G.; Wagner, E.F. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2002, 2, 389–406. [Google Scholar] [CrossRef]
- Veillette, C.J.; von Schroeder, H.P. Endothelin-1 down-regulates the expression of vascular endothelial growth factor-A associated with osteoprogenitor proliferation and differentiation. Bone 2004, 34, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Yue, J.; Liu, W. Broadening the role of osteocalcin in the hypothalamic-pituitary-gonadal axis. J. Endocrinol. 2021, 249, R43–R51. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Ghosh, A.; Guo, X.Z.; Wang, S.M.; Hou, Y.F.; Li, S.T.; Liu, J.M. Roles for osteocalcin in brain signalling: Implications in cognition- and motor-related disorders. Mol. Brain 2019, 12, 23. [Google Scholar] [CrossRef]
- Cantatore, F.P.; Crivellato, E.; Nico, B.; Ribatti, D. Osteocalcin is angiogenic in vivo. Cell Biol. Int. 2005, 29, 583–585. [Google Scholar] [CrossRef]
- Dou, J.; Li, H.; Ma, X.; Zhang, M.; Fang, Q.; Nie, M.; Bao, Y.; Jia, W. Osteocalcin attenuates high fat diet-induced impairment of endothelium-dependent relaxation through Akt/eNOS-dependent pathway. Cardiovasc. Diabetol. 2014, 13, 74. [Google Scholar] [CrossRef]
- Xu, R.; Yallowitz, A.; Qin, A.; Wu, Z.; Shin, D.Y.; Kim, J.M.; Debnath, S.; Ji, G.; Bostrom, M.P.; Yang, X.; et al. Targeting skeletal endothelium to ameliorate bone loss. Nat. Med. 2018, 24, 823–833. [Google Scholar] [CrossRef]
- Wang, H.; Yin, Y.; Li, W.; Zhao, X.; Yu, Y.; Zhu, J.; Qin, Z.; Wang, Q.; Wang, K.; Lu, W.; et al. Over-expression of PDGFR-beta promotes PDGF-induced proliferation, migration, and angiogenesis of EPCs through PI3K/Akt signaling pathway. PLoS ONE 2012, 7, e30503. [Google Scholar]
- Xie, H.; Cui, Z.; Wang, L.; Xia, Z.; Hu, Y.; Xian, L.; Li, C.; Xie, L.; Crane, J.; Wan, M.; et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat. Med. 2014, 20, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, L.; Liu, G.; Jandu, S.; Su, W.; Wodu, B.P.; Savage, W.; Poe, A.; Liu, X.; Alexander, L.M.; Cao, X.; et al. Skeleton-secreted PDGF-BB mediates arterial stiffening. J. Clin. Investig. 2021, 131, e147116. [Google Scholar] [CrossRef] [PubMed]
- Sang, Q.X. Complex role of matrix metalloproteinases in angiogenesis. Cell Res. 1998, 8, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front. Immunol. 2019, 10, 1278. [Google Scholar] [CrossRef] [PubMed]
- Carulli, C.; Innocenti, M.; Brandi, M.L. Bone vascularization in normal and disease conditions. Front. Endocrinol. 2013, 4, 106. [Google Scholar] [CrossRef]
- Lewin, E.; Olgaard, K. The vascular secret of Klotho. Kidney Int. 2015, 87, 1089–1091. [Google Scholar] [CrossRef]
- Block, G.A.; Hulbert-Shearon, T.E.; Levin, N.W.; Port, F.K. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am. J. Kidney Dis. 1998, 31, 607–617. [Google Scholar] [CrossRef]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, D.J.; Andress, D.L. Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 520–528. [Google Scholar] [CrossRef]
- Block, G.A.; Kilpatrick, R.D.; Lowe, K.A.; Wang, W.; Danese, M.D. CKD-mineral and bone disorder and risk of death and cardiovascular hospitalization in patients on hemodialysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 2132–2140. [Google Scholar] [CrossRef]
- Li, D.; Liu, W.; Huang, H.; Guo, W.; Diao, Z.; Chen, X.; Wangs, W. Association between the risk of death and serum calcium, phosphate, and intact parathyroid hormone levels in older patients undergoing maintenance hemodialysis: A cohort study in Beijing. Ther. Adv. Endocrinol. Metab. 2021, 12, 20420188211025161. [Google Scholar] [CrossRef]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Gao, Z.; Curhan, G. Cholesterol, Recurrent Events Trial, I. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005, 112, 2627–2633. [Google Scholar] [CrossRef]
- Dhingra, R.; Sullivan, L.M.; Fox, C.S.; Wang, T.J.; D’Agostino, R.B., Sr.; Gaziano, J.M.; Vasan, R.S. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch. Intern. Med. 2007, 167, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Slatopolsky, E.; Dusso, A.; Brown, A.J. The role of phosphorus in the development of secondary hyperparathyroidism and parathyroid cell proliferation in chronic renal failure. Am. J. Med. Sci. 1999, 317, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Mace, M.L.; Gravesen, E.; Nordholm, A.; Hofman-Bang, J.; Secher, T.; Olgaard, K.; Lewin, E. Kidney fibroblast growth factor 23 does not contribute to elevation of its circulating levels in uremia. Kidney Int. 2017, 92, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Abbasian, N.; Goodall, A.H.; Burton, J.O.; Bursnall, D.; Bevington, A.; Brunskill, N.J. Hyperphosphatemia Drives Procoagulant Microvesicle Generation in the Rat Partial Nephrectomy Model of CKD. J. Clin. Med. 2020, 9, 3534. [Google Scholar] [CrossRef]
- Naveh-Many, T.; Volovelsky, O. Parathyroid Cell Proliferation in Secondary Hyperparathyroidism of Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 4332. [Google Scholar] [CrossRef]
- Rukov, J.L.; Gravesen, E.; Mace, M.L.; Hofman-Bang, J.; Vinther, J.; Andersen, C.B.; Lewin, E.; Olgaard, K. Effect of chronic uremia on the transcriptional profile of the calcified aorta analysed by RNA-sequencing. Am. J. Physiol.-Ren. Physiol. 2016, 310, F477–F491. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146638 (accessed on 15 October 2021). [CrossRef]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Proudfoot, D.; Tyson, K.L.; Cary, N.R.; Edmonds, M.; Weissberg, P.L. Expression of mineralisation-regulating proteins in association with human vascular calcification. Z. Kardiol. 2000, 89 (Suppl. 2), 63–68. [Google Scholar] [CrossRef]
- Sanchis, P.; Ho, C.Y.; Liu, Y.; Beltran, L.E.; Ahmad, S.; Jacob, A.P.; Furmanik, M.; Laycock, J.; Long, D.A.; Shroff, R.; et al. Arterial "inflammaging" drives vascular calcification in children on dialysis. Kidney Int. 2019, 95, 958–972. [Google Scholar] [CrossRef]
- Zelt, J.G.; Svajger, B.A.; Quinn, K.; Turner, M.E.; Laverty, K.J.; Shum, B.; Holden, R.M.; Adams, M.A. Acute Tissue Mineral Deposition in Response to a Phosphate Pulse in Experimental CKD. J. Bone Miner. Res. 2019, 34, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Mace, M.L.; Olgaard, K.; Lewin, E. New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis. Int. J. Mol. Sci. 2020, 21, 8810. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Schurgers, L.J.; Shiels, P.G.; Stenvinkel, P. Early vascular ageing in chronic kidney disease: Impact of inflammation, vitamin K, senescence and genomic damage. Nephrol. Dial. Transplant. 2020, 35, ii31–ii37. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, N.C.; Cobo, G.; Dai, L.; Lindholm, B.; Stenvinkel, P. Role of Uremic Toxins in Early Vascular Ageing and Calcification. Toxins 2021, 13, 26. [Google Scholar] [CrossRef]
- Pasch, A.; Jahnen-Dechent, W.; Smith, E.R. Phosphate, Calcification in Blood, and Mineral Stress: The Physiologic Blood Mineral Buffering System and Its Association with Cardiovascular Risk. Int. J. Nephrol. 2018, 2018, 9182078. [Google Scholar] [CrossRef]
- Back, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceicao, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement From the COST Action EuroSoftCalcNet. Front. Cardiovasc. Med. 2018, 5, 196. [Google Scholar] [CrossRef]
- Gravesen, E.; Nordholm, A.; Mace, M.; Morevati, M.; Hogdall, E.; Nielsen, C.; Kjaer, A.; Olgaard, K.; Lewin, E. Effect of inhibition of CBP-coactivated beta-catenin-mediated Wnt signalling in uremic rats with vascular calcifications. PLoS ONE 2018, 13, e0201936. [Google Scholar] [CrossRef]
- Gravesen, E.; Lerche Mace, M.; Nordholm, A.; Hofman-Bang, J.; Hruska, K.; Haagen Nielsen, C.; Kjaer, A.; Olgaard, K.; Lewin, E. Exogenous BMP7 in aortae of rats with chronic uremia ameliorates expression of profibrotic genes, but does not reverse established vascular calcification. PLoS ONE 2018, 13, e0190820. [Google Scholar] [CrossRef]
- Nordholm, A.; Mace, M.L.; Gravesen, E.; Hofman-Bang, J.; Morevati, M.; Olgaard, K.; Lewin, E. Klotho and activin A in kidney injury: Plasma Klotho is maintained in unilateral obstruction despite no upregulation of Klotho biosynthesis in the contralateral kidney. Am. J. Physiol. Ren. Physiol. 2018, 314, F753–F762. [Google Scholar] [CrossRef]
- Mace, M.L.; Gravesen, E.; Nordholm, A.; Egstrand, S.; Morevati, M.; Nielsen, C.; Kjaer, A.; Behets, G.; D’Haese, P.; Olgaard, K.; et al. Chronic Kidney Disease-Induced Vascular Calcification Impairs Bone Metabolism. J. Bone Miner. Res. 2021, 36, 510–522. [Google Scholar] [CrossRef]
- Egstrand, S.; Mace, M.L.; Olgaard, K.; Lewin, E. The Vascular Circadian Clock in Chronic Kidney Disease. Cells 2021, 10, 1769. [Google Scholar] [CrossRef] [PubMed]
- Egstrand, S.; Nordholm, A.; Morevati, M.; Mace, M.L.; Hassan, A.; Naveh-Many, T.; Rukov, J.L.; Gravesen, E.; Olgaard, K.; Lewin, E. A molecular circadian clock operates in the parathyroid gland and is disturbed in chronic kidney disease associated bone and mineral disorder. Kidney Int. 2020, 98, 1461–1475. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Gilchrist, M.; Strain, D.; Fraser, D.; Shore, A. The systemic microcirculation in dialysis populations. Microcirculation 2020, 27, e12613. [Google Scholar] [CrossRef] [PubMed]
- Malyszko, J. Mechanism of endothelial dysfunction in chronic kidney disease. Clin. Chim. Acta. 2010, 411, 1412–1420. [Google Scholar] [CrossRef]
- Drueke, T.B.; Massy, Z.A. Atherosclerosis in CKD: Differences from the general population. Nat. Rev. Nephrol. 2010, 6, 723–735. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Rodriguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermudez-Lopez, M.; Sanchez-Nino, M.D.; Perez-Fernandez, M.; Ortiz, A. Atherosclerosis in Chronic Kidney Disease: More, Less, or Just Different? Arter. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef]
- Van den Bergh, G.; Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. The Vicious Cycle of Arterial Stiffness and Arterial Media Calcification. Trends Mol. Med. 2019, 25, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Badimon, L.; Borrell-Pages, M. Wnt signaling in the vessel wall. Curr. Opin. Hematol. 2017, 24, 230–239. [Google Scholar] [CrossRef]
- Hruska, K.A.; Sugatani, T.; Agapova, O.; Fang, Y. The chronic kidney disease—Mineral bone disorder (CKD-MBD): Advances in pathophysiology. Bone 2017, 100, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Cejka, D.; Herberth, J.; Branscum, A.J.; Fardo, D.W.; Monier-Faugere, M.C.; Diarra, D.; Haas, M.; Malluche, H.H. Sclerostin and Dickkopf-1 in renal osteodystrophy. Clin. J. Am. Soc. Nephrol. 2011, 6, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Behets, G.J.; Viaene, L.; Meijers, B.; Blocki, F.; Brandenburg, V.M.; Verhulst, A.; D’Haese, P.C.; Evenepoel, P. Circulating levels of sclerostin but not DKK1 associate with laboratory parameters of CKD-MBD. PLoS ONE 2017, 12, e0176411. [Google Scholar] [CrossRef]
- Zschiedrich, S.; Budde, K.; Nurnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Hoerl, W.H.; Obermuller, N.; Arns, W.; et al. Secreted frizzled-related protein 4 predicts progression of autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2016, 31, 284–289. [Google Scholar] [CrossRef][Green Version]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar] [CrossRef]
- Nordholm, A.; Egstrand, S.; Gravesen, E.; Mace, M.L.; Morevati, M.; Olgaard, K.; Lewin, E. Circadian rhythm of activin A and related parameters of mineral metabolism in normal and uremic rats. Pflügers Arch.-Eur. J. Physiol. 2019, 471, 1079–1094. [Google Scholar] [CrossRef]
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F.M. Activin A in Mammalian Physiology. Physiol. Rev. 2019, 99, 739–780. [Google Scholar] [CrossRef]
- Ling, N.; Ying, S.Y.; Ueno, N.; Shimasaki, S.; Esch, F.; Hotta, M.; Guillemin, R. Pituitary FSH is released by a heterodimer of the beta-subunits from the two forms of inhibin. Nature 1986, 321, 779–782. [Google Scholar] [CrossRef]
- Vale, W.; Rivier, J.; Vaughan, J.; McClintock, R.; Corrigan, A.; Woo, W.; Karr, D.; Spiess, J. Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature 1986, 321, 776–779. [Google Scholar] [CrossRef]
- Agapova, O.A.; Fang, Y.; Sugatani, T.; Seifert, M.E.; Hruska, K.A. Ligand trap for the activin type IIA receptor protects against vascular disease and renal fibrosis in mice with chronic kidney disease. Kidney Int. 2016, 89, 1231–1243. [Google Scholar] [CrossRef]
- Williams, M.J.; Sugatani, T.; Agapova, O.A.; Fang, Y.; Gaut, J.P.; Faugere, M.C.; Malluche, H.H.; Hruska, K.A. The activin receptor is stimulated in the skeleton, vasculature, heart, and kidney during chronic kidney disease. Kidney Int. 2018, 93, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Lodberg, A. Principles of the activin receptor signaling pathway and its inhibition. Cytokine Growth Factor Rev. 2021, 60, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ginsberg, C.; Seifert, M.; Agapova, O.; Sugatani, T.; Register, T.C.; Freedman, B.I.; Monier-Faugere, M.C.; Malluche, H.; Hruska, K.A. CKD-induced wingless/integration1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J. Am. Soc. Nephrol. 2014, 25, 1760–1773. [Google Scholar] [CrossRef]
- Egstrand, S.; Olgaard, K.; Lewin, E. Circadian rhythms of mineral metabolism in chronic kidney disease-mineral bone disorder. Curr. Opin. Nephrol. Hypertens. 2020, 29, 367–377. [Google Scholar] [CrossRef]
- Davies, M.R.; Lund, R.J.; Hruska, K.A. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J. Am. Soc. Nephrol. 2003, 14, 1559–1567. [Google Scholar] [CrossRef]
- Davies, M.R.; Lund, R.J.; Mathew, S.; Hruska, K.A. Low turnover osteodystrophy and vascular calcification are amenable to skeletal anabolism in an animal model of chronic kidney disease and the metabolic syndrome. J. Am. Soc. Nephrol. 2005, 16, 917–928. [Google Scholar] [CrossRef]
- Lewin, E.; Wang, W.; Olgaard, K. Reversibility of experimental secondary hyperparathyroidism. Kidney Int. 1997, 52, 1232–1241. [Google Scholar] [CrossRef][Green Version]
- Lewin, E.; Colstrup, H.; Pless, V.; Ladefoged, J.; Olgaard, K. A model of reversible uremia employing isogenic kidney transplantation in the rat. Reversibility of secondary hyperparathyroidism. Scand. J. Urol. Nephrol. 1993, 27, 115–120. [Google Scholar] [CrossRef]
- Brandenburg, V.M.; Kramann, R.; Koos, R.; Kruger, T.; Schurgers, L.; Muhlenbruch, G.; Hubner, S.; Gladziwa, U.; Drechsler, C.; Ketteler, M. Relationship between sclerostin and cardiovascular calcification in hemodialysis patients: A cross-sectional study. BMC Nephrol. 2013, 14, 219. [Google Scholar] [CrossRef]
- Morena, M.; Jaussent, I.; Dupuy, A.M.; Bargnoux, A.S.; Kuster, N.; Chenine, L.; Leray-Moragues, H.; Klouche, K.; Vernhet, H.; Canaud, B.; et al. Osteoprotegerin and sclerostin in chronic kidney disease prior to dialysis: Potential partners in vascular calcifications. Nephrol. Dial. Transplant. 2015, 30, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Guan, L.; Zhang, Y.; Yu, S.; Cao, B.; Ji, Y. Sclerostin as a new key factor in vascular calcification in chronic kidney disease stages 3 and 4. Int. Urol. Nephrol. 2016, 48, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Tong, D.; Ou, Y.; Zhang, H.; Zhang, Z.; Li, S.; Zhou, J.; Zhang, J.; Liao, E. Serum sclerostin levels were positively correlated with fat mass and bone mineral density in central south Chinese postmenopausal women. Clin. Endocrinol. 2012, 76, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Arasu, A.; Cawthon, P.M.; Lui, L.Y.; Do, T.P.; Arora, P.S.; Cauley, J.A.; Ensrud, K.E.; Cummings, S.R.; Study of Osteoporotic Fractures Research, G. Serum sclerostin and risk of hip fracture in older Caucasian women. J. Clin. Endocrinol. Metab. 2012, 97, 2027–2032. [Google Scholar] [CrossRef] [PubMed]
- Szulc, P.; Boutroy, S.; Vilayphiou, N.; Schoppet, M.; Rauner, M.; Chapurlat, R.; Hamann, C.; Hofbauer, L.C. Correlates of bone microarchitectural parameters and serum sclerostin levels in men: The STRAMBO study. J. Bone Miner. Res. 2013, 28, 1760–1770. [Google Scholar] [CrossRef]
- Thambiah, S.; Roplekar, R.; Manghat, P.; Fogelman, I.; Fraser, W.D.; Goldsmith, D.; Hampson, G. Circulating sclerostin and Dickkopf-1 (DKK1) in predialysis chronic kidney disease (CKD): Relationship with bone density and arterial stiffness. Calcif. Tissue Int. 2012, 90, 473–480. [Google Scholar] [CrossRef]
- Cejka, D.; Jager-Lansky, A.; Kieweg, H.; Weber, M.; Bieglmayer, C.; Haider, D.G.; Diarra, D.; Patsch, J.M.; Kainberger, F.; Bohle, B.; et al. Sclerostin serum levels correlate positively with bone mineral density and microarchitecture in haemodialysis patients. Nephrol. Dial. Transplant. 2012, 27, 226–230. [Google Scholar] [CrossRef]
- Pelletier, S.; Confavreux, C.B.; Haesebaert, J.; Guebre-Egziabher, F.; Bacchetta, J.; Carlier, M.C.; Chardon, L.; Laville, M.; Chapurlat, R.; London, G.M.; et al. Serum sclerostin: The missing link in the bone-vessel cross-talk in hemodialysis patients? Osteoporos. Int. 2015, 26, 2165–2174. [Google Scholar] [CrossRef]
- Ishimura, E.; Okuno, S.; Ichii, M.; Norimine, K.; Yamakawa, T.; Shoji, S.; Nishizawa, Y.; Inaba, M. Relationship between serum sclerostin, bone metabolism markers, and bone mineral density in maintenance hemodialysis patients. J. Clin. Endocrinol. Metab. 2014, 99, 4315–4320. [Google Scholar] [CrossRef]
- Kuo, T.H.; Lin, W.H.; Chao, J.Y.; Wu, A.B.; Tseng, C.C.; Chang, Y.T.; Liou, H.H.; Wang, M.C. Serum sclerostin levels are positively related to bone mineral density in peritoneal dialysis patients: A cross-sectional study. BMC Nephrol. 2019, 20, 266. [Google Scholar] [CrossRef]
- Dreyer, T.; Shah, M.; Doyle, C.; Greenslade, K.; Penney, M.; Creeke, P.; Kotian, A.; Ke, H.Z.; Naidoo, V.; Holdsworth, G. Recombinant sclerostin inhibits bone formation in vitro and in a mouse model of sclerosteosis. J. Orthop. Translat. 2021, 29, 134–142. [Google Scholar] [CrossRef] [PubMed]
- De Mare, A.; Maudsley, S.; Azmi, A.; Hendrickx, J.O.; Opdebeeck, B.; Neven, E.; D’Haese, P.C.; Verhulst, A. Sclerostin as Regulatory Molecule in Vascular Media Calcification and the Bone-Vascular Axis. Toxins 2019, 11, 428. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.S.; Murray, D.W.; Hurson, C.J.; O’Brien, J.; Doran, P.P.; O’Byrne, J.M. The role of Dkk1 in bone mass regulation: Correlating serum Dkk1 expression with bone mineral density. J. Orthop. Res. 2011, 29, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Fouda, N.; Abbas, A.A. Serum dickkopf-1 level in postmenopausal females: Correlation with bone mineral density and serum biochemical markers. J. Osteoporos. 2013, 2013, 460210. [Google Scholar] [CrossRef] [PubMed]
- Eijken, M.; Swagemakers, S.; Koedam, M.; Steenbergen, C.; Derkx, P.; Uitterlinden, A.G.; van der Spek, P.J.; Visser, J.A.; de Jong, F.H.; Pols, H.A.; et al. The activin A-follistatin system: Potent regulator of human extracellular matrix mineralization. FASEB J. 2007, 21, 2949–2960. [Google Scholar] [CrossRef] [PubMed]
- Persy, V.; D’Haese, P. Vascular calcification and bone disease: The calcification paradox. Trends Mol. Med. 2009, 15, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, Y.; Graciolli, F.G.; O’Brien, S.; Tang, W.; dos Reis, L.M.; Ryan, S.; Phillips, L.; Boulanger, J.; Song, W.; Bracken, C.; et al. Repression of osteocyte Wnt/beta-catenin signaling is an early event in the progression of renal osteodystrophy. J. Bone Miner. Res. 2012, 27, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef]
- Rowe, P.S. Regulation of bone-renal mineral and energy metabolism: The PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 61–86. [Google Scholar] [CrossRef]
- Epsley, S.; Tadros, S.; Farid, A.; Kargilis, D.; Mehta, S.; Rajapakse, C.S. The Effect of Inflammation on Bone. Front. Physiol. 2020, 11, 511799. [Google Scholar] [CrossRef]
- Graciolli, F.G.; Neves, K.R.; Barreto, F.; Barreto, D.V.; dos Reis, L.M.; Canziani, M.E.; Sabbagh, Y.; Carvalho, A.B.; Jorgetti, V.; Elias, R.M.; et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017, 91, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, I.M.H.; Saurbrey, S.A.K.; Hjortkjaer, H.O.; Brainin, P.; Carlson, N.; Ballegaard, E.L.F.; Kamper, A.L.; Christoffersen, C.; Feldt-Rasmussen, B.; Kofoed, K.F.; et al. Regional distribution and severity of arterial calcification in patients with chronic kidney disease stages 1-5: A cross-sectional study of the Copenhagen chronic kidney disease cohort. BMC Nephrol. 2020, 21, 534. [Google Scholar] [CrossRef] [PubMed]
- Ryan, Z.C.; Ketha, H.; McNulty, M.S.; McGee-Lawrence, M.; Craig, T.A.; Grande, J.P.; Westendorf, J.J.; Singh, R.J.; Kumar, R. Sclerostin alters serum vitamin D metabolite and fibroblast growth factor 23 concentrations and the urinary excretion of calcium. Proc. Natl. Acad. Sci. USA 2013, 110, 6199–6204. [Google Scholar] [CrossRef]
- Ito, N.; Prideaux, M.; Wijenayaka, A.R.; Yang, D.; Ormsby, R.T.; Bonewald, L.F.; Atkins, G.J. Sclerostin Directly Stimulates Osteocyte Synthesis of Fibroblast Growth Factor-23. Calcif. Tissue Int. 2021, 109, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Schuler, C.; Bergow, C.; Petric, A.; Erben, R.G. Augmented Fibroblast Growth Factor-23 Secretion in Bone Locally Contributes to Impaired Bone Mineralization in Chronic Kidney Disease in Mice. Front. Endocrinol. 2018, 9, 311. [Google Scholar] [CrossRef]
- Murali, S.K.; Roschger, P.; Zeitz, U.; Klaushofer, K.; Andrukhova, O.; Erben, R.G. FGF23 Regulates Bone Mineralization in a 1,25(OH) D and Klotho-Independent Manner. J. Bone Miner. Res. 2015, 1, 129–142. [Google Scholar]
- Carrillo-Lopez, N.; Panizo, S.; Alonso-Montes, C.; Roman-Garcia, P.; Rodriguez, I.; Martinez-Salgado, C.; Dusso, A.S.; Naves, M.; Cannata-Andia, J.B. Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int. 2016, 90, 77–89. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X.; Newman, C.L.; Organ, J.M.; Kneissel, M.; Kramer, I.; Gattone, V.H., 2nd; Allen, M.R. Anti-sclerostin antibody treatment in a rat model of progressive renal osteodystrophy. J. Bone Miner. Res. 2015, 30, 499–509. [Google Scholar] [CrossRef]
- Asci, G.; Ok, E.; Savas, R.; Ozkahya, M.; Duman, S.; Toz, H.; Kayikcioglu, M.; Branscum, A.J.; Monier-Faugere, M.C.; Herberth, J.; et al. The link between bone and coronary calcifications in CKD-5 patients on haemodialysis. Nephrol. Dial. Transplant. 2011, 26, 1010–1015. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Normal Aorta (Control) | CKD-Induced Calcified Aorta | Log2 Ratio CKD Aorta/Control | p-Value |
|---|---|---|---|---|
| Endothelial nitric oxide synthase (Nos3) | 7 | 6 | −0.17 | p = 0.65 |
| Prostaglandin (Ptgs2) | 27 | 40 | +0.53 | p = 0.02 |
| Endothelin 1 (Edn1) | 1.6 | 3.9 | +1.32 | p = 0.01 |
| Pecam (Pecam1) | 89 | 111 | +0.32 | p = 0.23 |
| RANK (Tnfrsf11a) | 2.1 | 3.2 | +0.56 | p = 1 |
| Osteoprotegerin (Tnfrsf11b) | 279 | 179 | −0.64 | p = 0.01 |
| BMP 2 (Bmp2) | 1.6 | 3.0 | +0.89 | p = 1 |
| Runt-related transcription factor 2 (Runx2) | 1.3 | 6.0 | +2.18 | p < 0.001 |
| Noggin (Nog) | 0.25 | 1.2 | +2.24 | p = 1 |
| Pleiotrophin (Ptn) | 7 | 13 | +0.78 | p = 0.001 |
| VEGF-A (Vegfa) | 112 | 132 | +0.24 | p = 0.46 |
| Osteopontin (Spp1) | 443 | 6552 | +3.88 | p < 0.001 |
| MMP 2 (Mmp2) | 230 | 240 | +0.06 | p = 0.90 |
| Slit homolog protein 3 (Slit3) | 61 | 116 | +0.93 | p < 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mace, M.L.; Egstrand, S.; Morevati, M.; Olgaard, K.; Lewin, E. New Insights to the Crosstalk between Vascular and Bone Tissue in Chronic Kidney Disease–Mineral and Bone Disorder. Metabolites 2021, 11, 849. https://doi.org/10.3390/metabo11120849
Mace ML, Egstrand S, Morevati M, Olgaard K, Lewin E. New Insights to the Crosstalk between Vascular and Bone Tissue in Chronic Kidney Disease–Mineral and Bone Disorder. Metabolites. 2021; 11(12):849. https://doi.org/10.3390/metabo11120849
Chicago/Turabian StyleMace, Maria L., Søren Egstrand, Marya Morevati, Klaus Olgaard, and Ewa Lewin. 2021. "New Insights to the Crosstalk between Vascular and Bone Tissue in Chronic Kidney Disease–Mineral and Bone Disorder" Metabolites 11, no. 12: 849. https://doi.org/10.3390/metabo11120849
APA StyleMace, M. L., Egstrand, S., Morevati, M., Olgaard, K., & Lewin, E. (2021). New Insights to the Crosstalk between Vascular and Bone Tissue in Chronic Kidney Disease–Mineral and Bone Disorder. Metabolites, 11(12), 849. https://doi.org/10.3390/metabo11120849