
Clinical Insights into Mitochondrial Neurodevelopmental and Neurodegenerative Disorders: Their Biosignatures from Mass Spectrometry-Based Metabolomics


Abstract
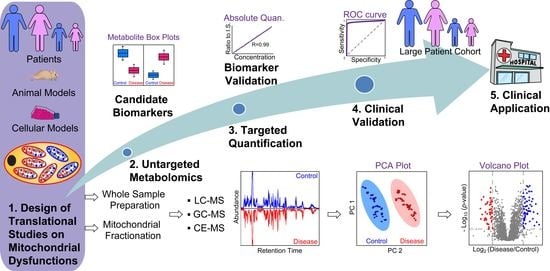
:
1. Introduction
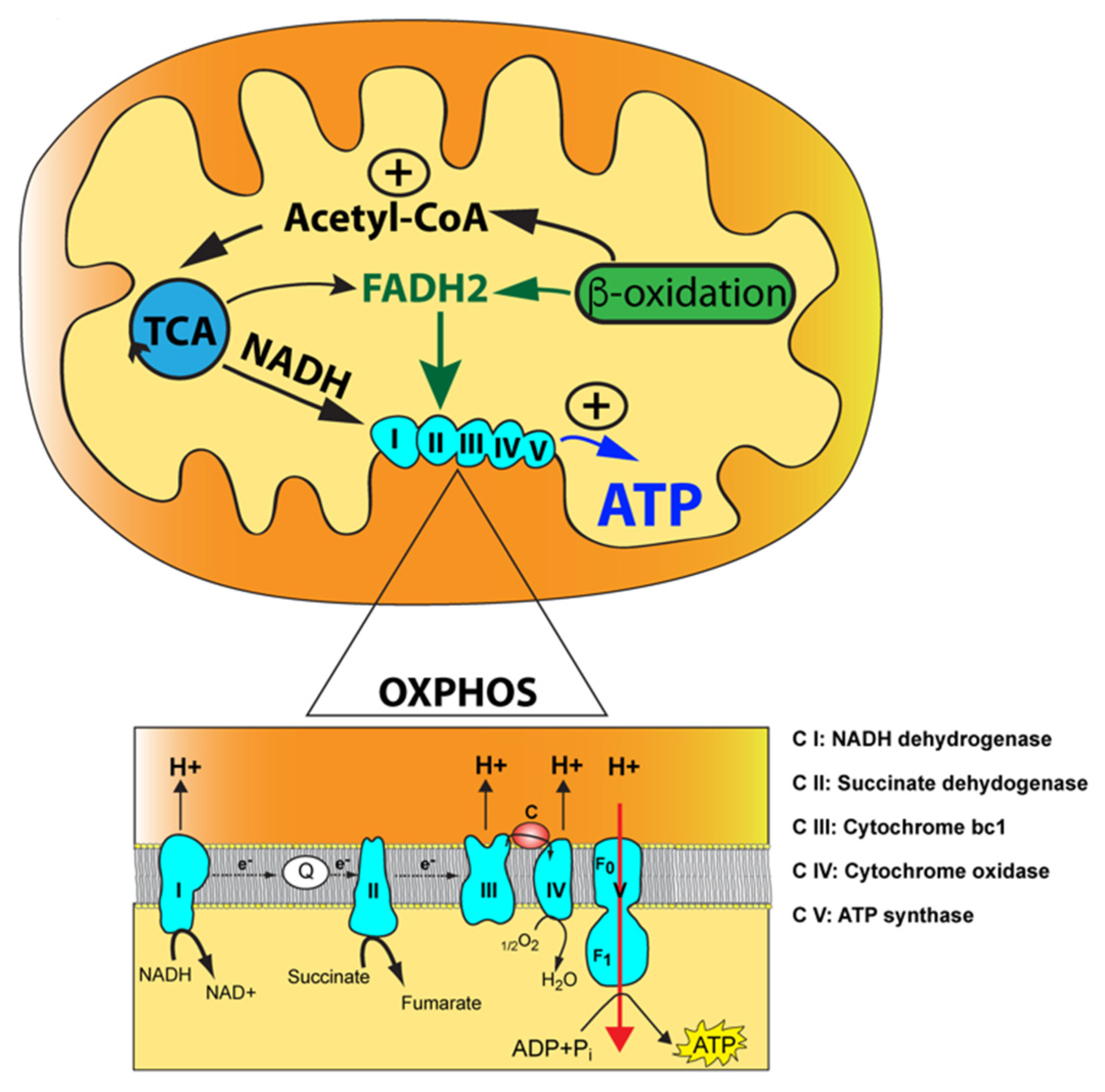
1.1. Mitochondria Function as Metabolic Nodes
1.2. Mitochondrial Inheritance
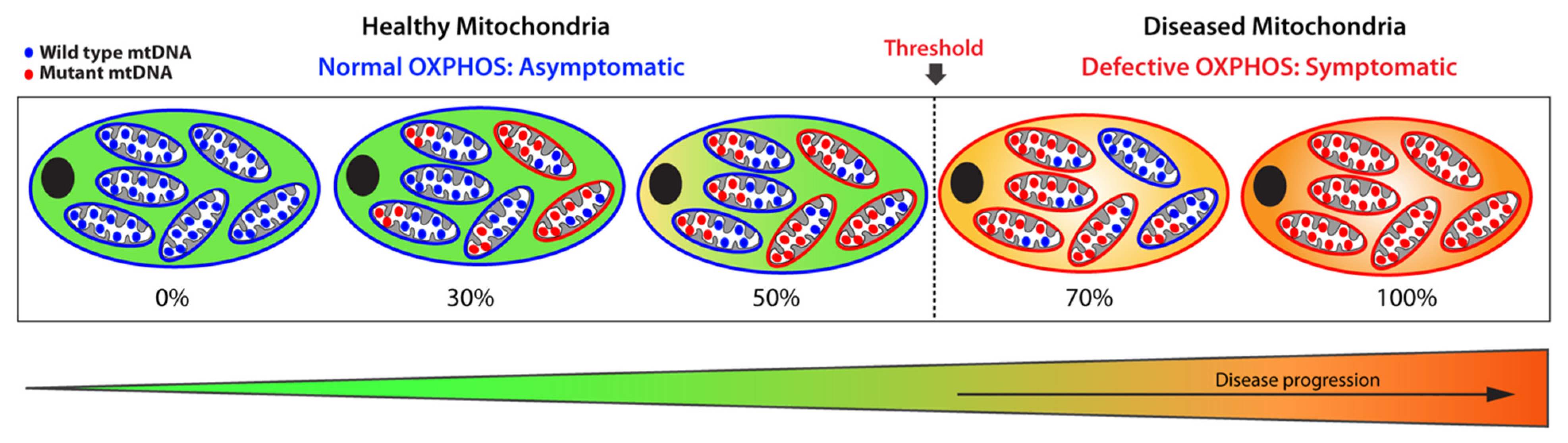
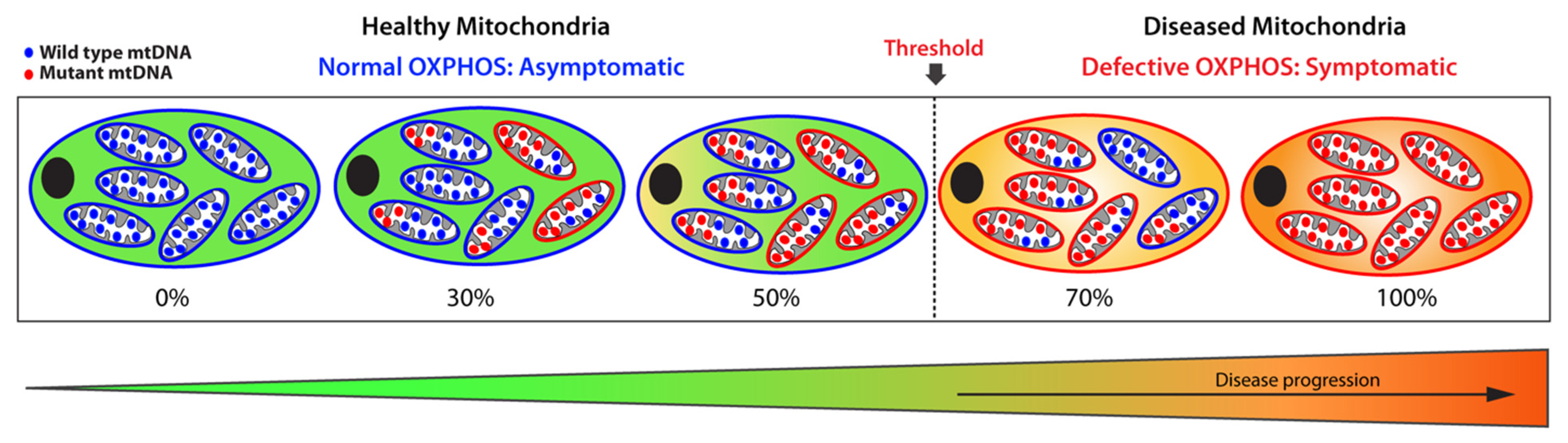
1.3. Mitochondrial Copy Number and Heteroplasmy
2. Genetics of Mitochondrial Diseases
2.1. Primary Mitochondrial Diseases
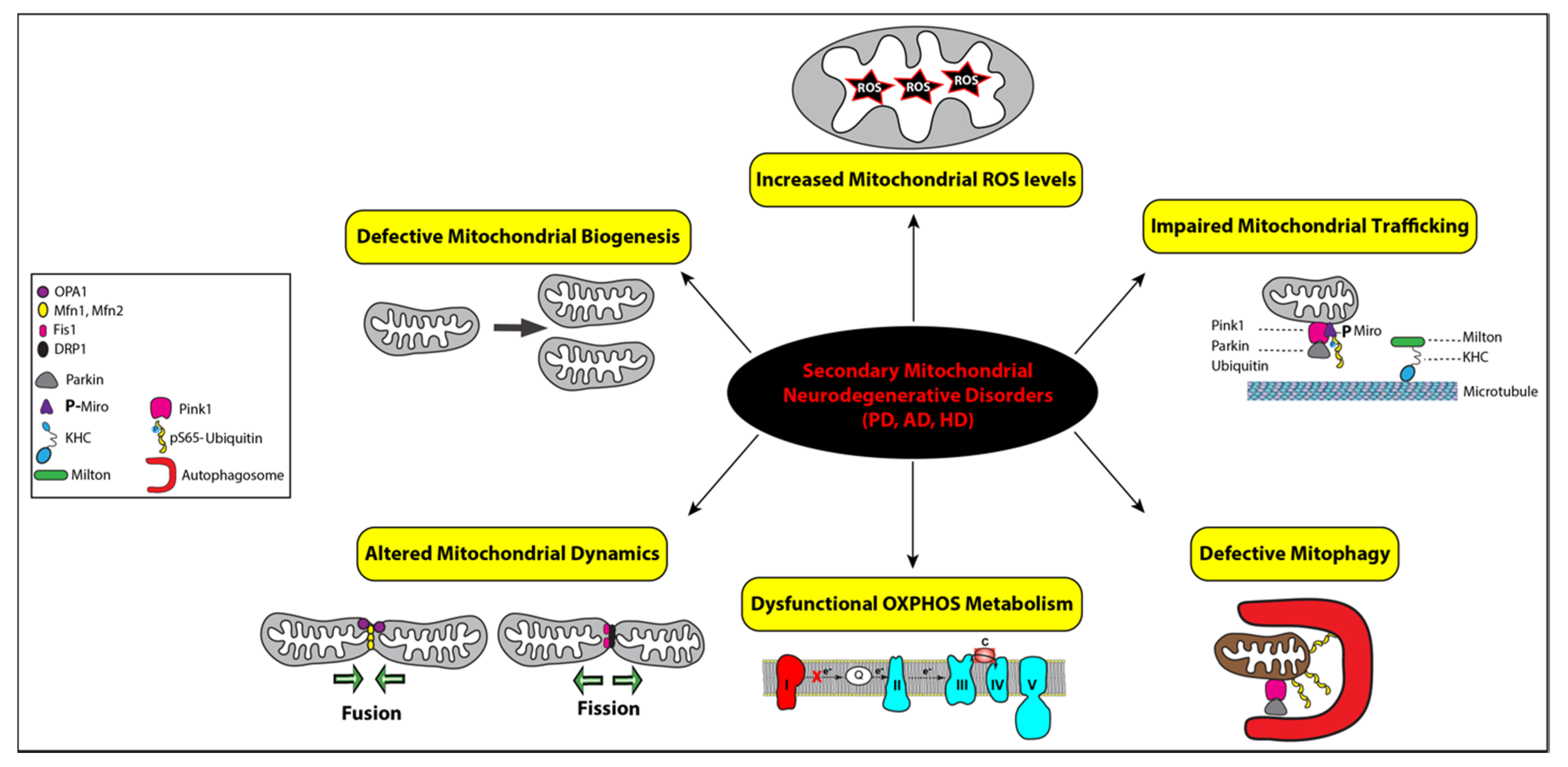
2.2. Secondary Mitochondrial Diseases
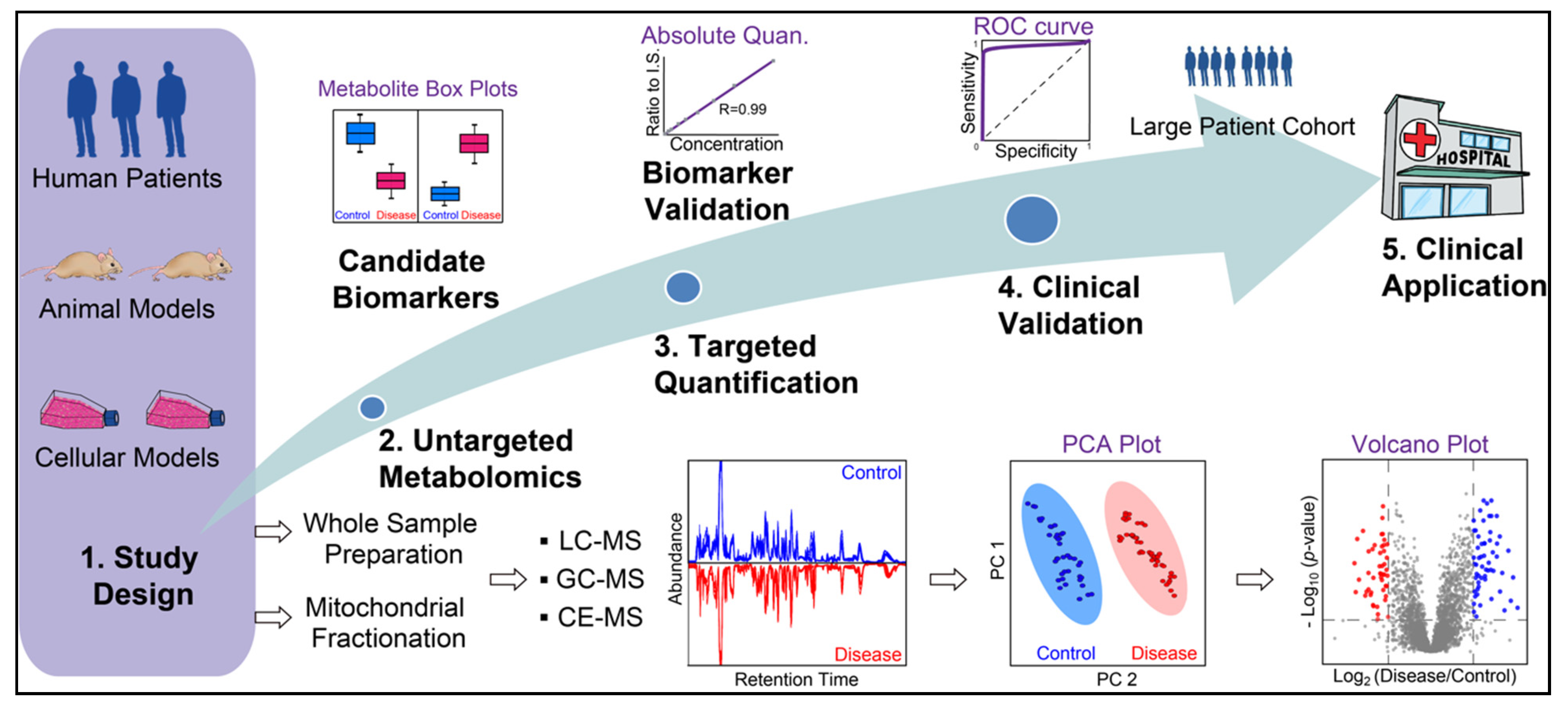
3. MS-Based Metabolomics to Study Mitochondrial Disease
3.1. Introduction of MS-Based Metabolomics
3.2. Mitochondrial Metabolomics
3.3. MS-Based Metabolomics in Primary Mitochondrial Diseases
3.4. MS-Based Metabolomics in Secondary Mitochondrial Disease
3.5. Metabolomics in Mitochondrial Disease-Specific Induced Pluripotent Stem Cells (iPSCs)
4. Conclusions and Future Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [Green Version]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Wallace, D.C. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu. Rev. Biochem. 2007, 76, 781–821. [Google Scholar] [CrossRef] [Green Version]
- Sherengul, W.; Kondo, R.; Matsuura, E.T. Analysis of paternal transmission of mitochondrial DNA in Drosophila. Genes Genet. Syst. 2006, 81, 399–404. [Google Scholar] [CrossRef] [Green Version]
- Nunes, M.D.S.; Dolezal, M.; Schlötterer, C. Extensive paternal mtDNA leakage in natural populations of Drosophila melanogaster. Mol. Ecol. 2013, 22, 2106–2117. [Google Scholar] [CrossRef] [Green Version]
- Gyllensten, U.; Wharton, D.; Josefsson, A.; Wilson, A.C. Paternal inheritance of mitochondrial DNA in mice. Nature 1991, 352, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, N.; Guo, W.; Hu, X.; Liu, Z.; Gong, G.; Wang, A.; Feng, J.; Wu, C. Further evidence for paternal inheritance of mitochondrial DNA in the sheep (Ovis aries). Heredity 2004, 93, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, H.; Li, H.; Nakagawa, A.; Lin, J.L.J.; Lee, E.S.; Harry, B.L.; Skeen-Gaar, R.R.; Suehiro, Y.; William, D.; et al. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science 2016, 353, 394–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.R. Inheritance: Male mtDNA Just Can’t Catch a Break. Curr. Biol. 2017, 27, R264–R266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; O’Farrell, P.H.; Yakubovich, N.; DeLuca, S.Z. The Mitochondrial DNA Polymerase Promotes Elimination of Paternal Mitochondrial Genomes. Curr. Biol. 2017, 27, 1033–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.H.; Yi, Y.J.; Sutovsky, M.; Meyers, S.; Sutovsky, P. Autophagy and ubiquitin-proteasome system contribute to sperm mitophagy after mammalian fertilization. Proc. Natl. Acad. Sci. USA 2016, 113, E5261–E5270. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental inheritance of mitochondrial DNA in humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladoukakis, E.D.; Eyre-Walker, A. Evolutionary genetics: Direct evidence of recombination in human mitochondrial DNA. Heredity 2004, 93–321. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [Green Version]
- Marlow, F.L. Mitochondrial matters: Mitochondrial bottlenecks, self-assembling structures, and entrapment in the female germline. Stem Cell Res. 2017, 21, 178–186. [Google Scholar] [CrossRef]
- Al Rawi, S.; Louvet-Vallée, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Sharpley, M.S.; Marciniak, C.; Eckel-Mahan, K.; McManus, M.; Crimi, M.; Waymire, K.; Lin, C.S.; Masubuchi, S.; Friend, N.; Koike, M.; et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 2012, 151, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Ao, A.; Zhang, X.; Cyr, D.; Dufort, D.; Shoubridge, E.A. The role of mitochondrial DNA copy number in mammalian fertility. Biol. Reprod. 2010, 83, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Larsson, N.G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef] [Green Version]
- Alexeyev, M.F. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 2009, 276, 5768–5787. [Google Scholar] [CrossRef] [Green Version]
- Uittenbogaard, M.; Wang, H.; Zhang, V.W.; Wong, L.J.; Brantner, C.A.; Gropman, A.; Chiaramello, A. The nuclear background influences the penetrance of the near-homoplasmic m.1630 A > G MELAS variant in a symptomatic proband and asymptomatic mother. Mol. Genet. Metab. 2019, 126, 429–438. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Chiaramello, A. Maternally inherited mitochondrial respiratory disorders: From pathogenetic principles to therapeutic implications. Mol. Genet. Metab. 2020, 131, 38–52. [Google Scholar] [CrossRef]
- Wong, L.J.C. Challenges of Bringing Next Generation Sequencing Technologies to Clinical Molecular Diagnostic Laboratories. Neurotherapeutics 2013, 10, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, B.A.I.; Wilson, I.J.; Yu-Wai-Man, P.; Coxhead, J.; Deehan, D.; Horvath, R.; Taylor, R.W.; Samuels, D.C.; Santibanez-Koref, M.; Chinnery, P.F. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 2013, 22, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndr. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Suomalainen, A. Mitochondrial roles in disease: A box full of surprises. EMBO Mol. Med. 2015, 7, 1245–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Schon, K.R.; Ratnaike, T.; van den Ameele, J.; Horvath, R.; Chinnery, P.F. Mitochondrial Diseases: A Diagnostic Revolution. Trends Genet. 2020, 36, 702–717. [Google Scholar] [CrossRef]
- Man, P.Y.W.; Turnbull, D.M.; Chinnery, P.F. Leber hereditary optic neuropathy. J. Med. Genet. 2002, 39, 162–169. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- KEARNS, T.P.; SAYRE, G.P. Retinitis pigmentosa, external ophthalmoplegia, and complete heart block: Unusual syndrome with histologic study in one of two cases. AMA Arch. Ophthalmol. 1958, 60, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, S.G.; Phillips, P.C.; DiMauro, S.; De Vivo, D.C.; Rowland, L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984, 16, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Glatz, C.; D’Aco, K.; Smith, S.; Sondheimer, N. Mutation in the mitochondrial tRNA Val causes mitochondrial encephalopathy, lactic acidosis and stroke-like episodes. Mitochondrion 2011, 11, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Moraes, C.T.; Ciacci, F.; Silvestri, G.; Shanske, S.; Sciacco, M.; Hirano, M.; Schon, E.A.; Bonilla, E.; DiMauro, S. Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromuscul. Disord. 1993, 3, 43–50. [Google Scholar] [CrossRef]
- Ravn, K.; Wibrand, F.; Hansen, F.J.; Horn, N.; Rosenberg, T.; Schwartz, M. An mtDNA mutation, 14453GτA, in the NADH dehydrogenase subunit 6 associated with severe MELAS syndrome. Eur. J. Hum. Genet. 2001, 9, 805–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Tatuch, Y.; Christodoulou, J.; Feigenbaum, A.; Clarke, J.T.R.; Wherret, J.; Smith, C.; Rudd, N.; Petrova-Benedict, R.; Robinson, B.H. Heteroplasmic mtDNA mutation (T→G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am. J. Hum. Genet. 1992, 50, 852–858. [Google Scholar]
- Uittenbogaard, M.; Brantner, C.A.; Fang, Z.S.; Wong, L.J.C.; Gropman, A.; Chiaramello, A. Novel insights into the functional metabolic impact of an apparent de novo m.8993T>G variant in the MT-ATP6 gene associated with maternally inherited form of Leigh Syndrome. Mol. Genet. Metab. 2018, 124, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Blok, R.B.; Dahl, H.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, R.S.; LeWitt, P.A.; Ebert, M.H.; Pakkenberg, H.; Kopin, I.J. The clinical syndrome of striatal dopamine deficiency. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). N. Engl. J. Med. 1985, 312, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [Green Version]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Zaltieri, M.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P.; Bellucci, A. Mitochondrial dysfunction and α-synuclein synaptic pathology in Parkinson’s disease: Who’s on first? Parkinsons Dis. 2015, 2015, 108029. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St. Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.H.; Li, L.; Vanhauwaert, R.; Nguyen, K.T.; Davis, M.D.; Bu, G.; Wszolek, Z.K.; Wang, X. Miro1 Marks Parkinson’s Disease Subset and Miro1 Reducer Rescues Neuron Loss in Parkinson’s Models. Cell Metab. 2019, 30, 1131–1140.e7. [Google Scholar] [CrossRef]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; Lavoie, M.J.; Schwarz, T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Singh, S. Updates on immunity and inflammation in Parkinson disease pathology. J. Neurosci. Res. 2018, 96, 379–390. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Spuch, C.; Ortolano, S.; Navarro, C. New insights in the amyloid-beta interaction with mitochondria. J. Aging Res. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Oliver, D.M.A. Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Kish, S.J. Mitochondrial Function and Dysfunction. Int. Rev. Neurobiol. 2002, 53, 341–385. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Hirai, K.; Hsiao, K.; Pappolla, M.A.; Harris, P.L.R.; Siedlak, S.L.; Tabaton, M.; Perry, G. Amyloid-β Deposition in Alzheimer Transgenic Mice Is Associated with Oxidative Stress. J. Neurochem. 2002, 70, 2212–2215. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Lah, J.J.; Levey, A.I.; Cutler, D.J. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol. 2012, 69, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; Strooper, B. De The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 1–15. [Google Scholar] [CrossRef]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.; Sims, R.; Leonenko, G.; Frizzati, A.; Harwood, J.C.; Grozeva, D.; Morgan, K.; Passmore, P.; Holmes, C.; Powell, J.; et al. Gene-based analysis in HRC imputed genome wide association data identifies three novel genes for Alzheimer’s disease. PLoS ONE 2019, 14, e0218111. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef]
- Choo, Y.S.; Johnson, G.V.W.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, B.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing p53 Network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef]
- Havelund, J.F.; Heegaard, N.H.H.; Færgeman, N.J.K.; Gramsbergen, J.B. Biomarker research in parkinson’s disease using metabolite profiling. Metabolites 2017, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, M.S.; Carvalho, M.; Bastos, M.L.; Guedes de Pinho, P. Metabolomics Analysis for Biomarker Discovery: Advances and Challenges. Curr. Med. Chem. 2013, 20, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Tran, T.; Zheng, J.; Sapkota, S.; MacDonald, S.W.; Camicioli, R.; Dixon, R.A.; Li, L. Metabolomics Analyses of Saliva Detect Novel Biomarkers of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 65, 1401–1416. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Greer, T.; Page, D.; Shi, Y.; Vezina, C.M.; Macoska, J.A.; Marker, P.C.; Bjorling, D.E.; Bushman, W.; Ricke, W.A.; et al. In-depth characterization and validation of human urine metabolomes reveal novel metabolic signatures of lower urinary tract symptoms. Sci. Rep. 2016, 6, 30869. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Wang, J.; Page, D.; Asthana, S.; Zetterberg, H.; Carlsson, C.; Okonkwo, O.C.; Li, L. Comparative Evaluation of MS-based Metabolomics Software and Its Application to Preclinical Alzheimer’s Disease. Sci. Rep. 2018, 8, 9291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Hao, L.; Thomas, S.; Buchberger, A.R.; Steinke, L.; Marker, P.C.; Ricke, W.A.; Li, L. Urinary Amine Metabolomics Characterization with Custom 12-Plex Isobaric DiLeu Labeling. J. Am. Soc. Mass Spectrom. 2020, 31, 1854–1860. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M.; Kwock, L.; Green, C. MELAS syndrome: Imaging and proton MR spectroscopic findings. Am. J. Neuroradiol. 1995, 16, 233–239. [Google Scholar] [PubMed]
- Möller, H.E.; Kurlemann, G.; Pützler, M.; Wiedermann, D.; Hilbich, T.; Fiedler, B. Magnetic resonance spectroscopy in patients with MELAS. J. Neurol. Sci. 2005, 229–230, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.Y.; Zhuang, Q.Q.; Qiu, Y.; Zhu, X.F.; Yan, Q.F. Cell models and drug discovery for mitochondrial diseases. J. Zhejiang Univ. Sci. B 2019, 20, 449–456. [Google Scholar] [CrossRef]
- Frankenfield, A.M.; Fernandopulle, M.S.; Hasan, S.; Ward, M.E.; Hao, L. Development and Comparative Evaluation of Endolysosomal Proximity Labeling-Based Proteomic Methods in Human iPSC-Derived Neurons. Anal. Chem. 2020, 92. [Google Scholar] [CrossRef]
- Zhong, X.; Hao, L.; Lu, J.; Ye, H.; Zhang, S.C.; Li, L. Quantitative analysis of serotonin secreted by human embryonic stem cells-derived serotonergic neurons via pH-mediated online stacking-CE-ESI-MRM. Electrophoresis 2016, 37, 1027–1030. [Google Scholar] [CrossRef]
- Esterhuizen, K.; van der Westhuizen, F.H.; Louw, R. Metabolomics of mitochondrial disease. Mitochondrion 2017, 35, 97–110. [Google Scholar] [CrossRef]
- Kuehnbaum, N.L.; Britz-McKibbin, P. New Advances in Separation Science for Metabolomics: Resolving Chemical Diversity in a Post-Genomic Era. Chem. Rev. 2013, 113, 2437–2468. [Google Scholar] [CrossRef]
- Ramautar, R.; Somsen, G.W.; de Jong, G.J. CE-MS for metabolomics: Developments and applications in the period 2010–2012. Electrophoresis 2013, 34, 86–98. [Google Scholar] [CrossRef]
- Hao, L.; Zhong, X.; Greer, T.; Ye, H.; Li, L. Relative quantification of amine-containing metabolites using isobaric N,N-dimethyl leucine (DiLeu) reagents via LC-ESI-MS/MS and CE-ESI-MS/MS. Analyst 2015, 140, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Siuzdak, G. Bioinformatics: The next frontier of metabolomics. Anal. Chem. 2015, 87, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, M.; Kawakami, M.; Robert, M.; Soga, T.; Tomita, M. Bioinformatics Tools for Mass Spectroscopy-Based Metabolomic Data Processing and Analysis. Curr. Bioinform. 2012, 7, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Koal, T.; Wang, Y.; Kohl, M.; Enot, D.P.; Deigner, H.P. Targeted metabolomics for biomarker discovery. Angew. Chem. Int. Ed. 2010, 49, 5426–5445. [Google Scholar] [CrossRef] [PubMed]
- Stauber, J.; Lemaire, R.; Franck, J.; Bonnel, D.; Croix, D.; Day, R.; Wisztorski, M.; Fournier, I.; Salzet, M. MALDI imaging of formalin-fixed paraffin-embedded tissues: Application to model animals of parkinson disease for biomarker hunting. J. Proteome Res. 2008, 7, 969–978. [Google Scholar] [CrossRef] [Green Version]
- Pierson, J.; Norris, J.L.; Aerni, H.R.; Svenningsson, P.; Caprioli, R.M.; Andrén, P.E. Molecular Profiling of Experimental Parkinson’s Disease: Direct Analysis of Peptides and Proteins on Brain Tissue Sections by MALDI Mass Spectrometry. J. Proteome Res. 2004, 3, 289–295. [Google Scholar] [CrossRef]
- Stoeckli, M.; Staab, D.; Staufenbiel, M.; Wiederhold, K.H.; Signor, L. Molecular imaging of amyloid β peptides in mouse brain sections using mass spectrometry. Anal. Biochem. 2002, 311, 33–39. [Google Scholar] [CrossRef]
- Matsumoto, J.; Sugiura, Y.; Yuki, D.; Hayasaka, T.; Goto-Inoue, N.; Zaima, N.; Kunii, Y.; Wada, A.; Yang, Q.; Nishiura, K.; et al. Abnormal phospholipids distribution in the prefrontal cortex from a patient with schizophrenia revealed by matrix-assisted laser desorption/ionization imaging mass spectrometry. Anal. Bioanal. Chem. 2011, 400, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Acquadro, E.; Caron, I.; Tortarolo, M.; Bucci, E.M.; Bendotti, C.; Corpillo, D. Human SOD1-G93A specific distribution evidenced in murine brain of a transgenic model for amyotrophic lateral sclerosis by MALDI imaging mass spectrometry. J. Proteome Res. 2014, 13, 1800–1809. [Google Scholar] [CrossRef]
- Angelini, R.; Vitale, R.; Patil, V.A.; Cocco, T.; Ludwig, B.; Greenberg, M.L.; Corcelli, A. Lipidomics of intact mitochondria by MALDI-TOF/MS. J. Lipid Res. 2012, 53, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Boggio, K.J.; Obasuyi, E.; Sugino, K.; Nelson, S.B.; Agar, N.Y.R.; Agar, J.N. Recent advances in single-cell MALDI mass spectrometry imaging and potential clinical impact. Expert Rev. Proteom. 2012, 8, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, Y.; Zaima, N.; Setou, M.; Ito, S.; Yao, I. Visualization of acetylcholine distribution in central nervous system tissue sections by tandem imaging mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1851–1861. [Google Scholar] [CrossRef] [Green Version]
- Gemperline, E.; Chen, B.; Li, L. Challenges and recent advances in mass spectrometric imaging of neurotransmitters. Bioanalysis 2014, 6, 525–540. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Wang, Y.; Chen, B.; Ma, F.; Hao, L.; Li, G.; Ouyang, C.; Li, L. Visualization and Identification of Neurotransmitters in Crustacean Brain via Multifaceted Mass Spectrometric Approaches. ACS Chem. Neurosci. 2019, 10. [Google Scholar] [CrossRef]
- Jiang, S.; Liang, Z.; Hao, L.; Li, L. Investigation of signaling molecules and metabolites found in crustacean hemolymph via in vivo microdialysis using a multifaceted mass spectrometric platform. Electrophoresis 2016, 37, 1031–1038. [Google Scholar] [CrossRef] [Green Version]
- Li, N.J.; Liu, W.T.; Li, W.; Li, S.Q.; Chen, X.H.; Bi, K.S.; He, P. Plasma metabolic profiling of Alzheimer’s disease by liquid chromatography/mass spectrometry. Clin. Biochem. 2010, 43, 992–997. [Google Scholar] [CrossRef]
- Haxby, J.V.; Grady, C.L.; Friedland, R.P.; Rapoport, S.I. Neocortical metabolic abnormalities precede nonmemory cognitive impairments in early dementia of the Alzheimer type: Longitudinal confirmation. J. Neural Transm. Suppl. 1987, 24, 49–53. [Google Scholar] [PubMed]
- Maurer, I.; Zierz, S.; Möller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Peisl, B.Y.L.; Schymanski, E.L.; Wilmes, P. Dark matter in host-microbiome metabolomics: Tackling the unknowns–A review. Anal. Chim. Acta 2018, 1037, 13–27. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.W.; Freinkman, E.; Sabatini, D.M. Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat. Protoc. 2017, 12, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- HOGEBOOM, G.H. Isolation of inatct mitochondria from rat liver; some biochemical properties of mitochondria and submicroscopic particulate material. J. Biol. Chem. 1947, 177, 847–858. [Google Scholar] [CrossRef]
- Matuszczyk, J.C.; Teleki, A.; Pfizenmaier, J.; Takors, R. Compartment-specific metabolomics for CHO reveals that ATP pools in mitochondria are much lower than in cytosol. Biotechnol. J. 2015, 10, 1639–1650. [Google Scholar] [CrossRef]
- Bowsher, C.G.; Tobin, A.K. Compartmentation of metabolism within mitochondria and plastids. J. Exp. Bot. 2001, 52, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337.e11. [Google Scholar] [CrossRef] [PubMed]
- Corcelli, A.; Saponetti, M.S.; Zaccagnino, P.; Lopalco, P.; Mastrodonato, M.; Liquori, G.E.; Lorusso, M. Mitochondria isolated in nearly isotonic KCl buffer: Focus on cardiolipin and organelle morphology. Biochim. Biophys. Acta Biomembr. 2010, 1798, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Bayraktar, E.C.; Baudrier, L.; Özerdem, C.; Lewis, C.A.; Chan, S.H.; Kunchok, T.; Abu-Remaileh, M.; Cangelosi, A.L.; Sabatini, D.M.; Birsoy, K.; et al. MITO-Tag Mice enable rapid isolation and multimodal profiling of mitochondria from specific cell types in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Sauer, U. Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2006, 2, 62. [Google Scholar] [CrossRef] [Green Version]
- Llufrio, E.M.; Cho, K.; Patti, G.J. Systems-level analysis of isotopic labeling in untargeted metabolomic data by X(13)CMS. Nat. Protoc. 2019, 14, 1970–1990. [Google Scholar] [CrossRef]
- Lee, W.D.; Mukha, D.; Aizenshtein, E.; Shlomi, T. Spatial-fluxomics provides a subcellular-compartmentalized view of reductive glutamine metabolism in cancer cells. Nat. Commun. 2019, 10, 1351. [Google Scholar] [CrossRef] [Green Version]
- Nikkanen, J.; Forsström, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamäki, J.; Roivainen, A.; Marjamäki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 2017, 6, e30952. [Google Scholar] [CrossRef] [PubMed]
- Schober, F.A.; Moore, D.; Atanassov, I.; Moedas, M.F.; Clemente, P.; Végvári, Á.; Fissi, N.E.; Filograna, R.; Bucher, A.-L.; Hinze, Y.; et al. The one-carbon pool controls mitochondrial energy metabolism via complex I and iron-sulfur clusters. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Scolamiero, E.; Cozzolino, C.; Albano, L.; Ansalone, A.; Caterino, M.; Corbo, G.; Di Girolamo, M.G.; Di Stefano, C.; Durante, A.; Franzese, G.; et al. Targeted metabolomics in the expanded newborn screening for inborn errors of metabolism. Mol. Biosyst. 2015, 11, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Chao De La Barca, J.M.; Simard, G.; Amati-Bonneau, P.; Safiedeen, Z.; Prunier-Mirebeau, D.; Chupin, S.; Gadras, C.; Tessier, L.; Gueguen, N.; Chevrollier, A.; et al. The metabolomic signature of Leber’s hereditary optic neuropathy reveals endoplasmic reticulum stress. Brain 2016, 139, 2864–2876. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Liu, J.; Zhou, J.; Liang, H.; Zhu, Y.; Wang, Q.; Leng, Y.; Zhang, Z.; Yuan, Y.; Wang, Z.; et al. Lipidomic profiling of plasma samples from patients with mitochondrial disease. Biochem. Biophys. Res. Commun. 2018, 500, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Cuillerier, A.; Daneault, C.; Deschênes, S.; Frayne, I.R.; Bouchard, B.; Forest, A.; Legault, J.T.; Vaz, F.M.; Rioux, J.D.; et al. Lipidomics unveils lipid dyshomeostasis and low circulating plasmalogens as biomarkers in a monogenic mitochondrial disorder. JCI Insight 2019, 4, 14. [Google Scholar] [CrossRef]
- Naviaux, R.K. Metabolic features of the cell danger response. Mitochondrion 2014, 16, 7–17. [Google Scholar] [CrossRef]
- Tanaka, M.; Nishigaki, Y.; Fuku, N.; Ibi, T.; Sahashi, K.; Koga, Y. Therapeutic potential of pyruvate therapy for mitochondrial diseases. Mitochondrion 2007, 7, 399–401. [Google Scholar] [CrossRef]
- Kami, K.; Fujita, Y.; Igarashi, S.; Koike, S.; Sugawara, S.; Ikeda, S.; Sato, N.; Ito, M.; Tanaka, M.; Tomita, M.; et al. Metabolomic profiling rationalized pyruvate efficacy in cybrid cells harboring MELAS mitochondrial DNA mutations. Mitochondrion 2012, 12, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Hamazaki, T.; Kudo, S.; Shintaku, H. Metabolic Signature of MELAS/Leigh Overlap Syndrome in Patient-specific Induced Pluripotent Stem Cells Model. Osaka City Med. J. 2016, 62, 69–76. [Google Scholar]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S. Emerging therapeutic roles for NAD + metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 2016, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voet, D.; Voet, J.G. Biochemistry, 2nd ed.; Wiley: New York, NY, USA, 1995; Volume 613, p. 21205. [Google Scholar]
- Munnich, A.; Rötig, A.; Chretien, D.; Cormier, V.; Bourgeron, T.; Bonnefont, J.P.; Saudubray, J.M.; Rustin, P. Clinical presentation of mitochondrial disorders in childhood. J. Inherit. Metab. Dis. 1996, 19, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Munnich, A.; Rötig, A.; Chretien, D.; Saudubray, J.M.; Cormier, V.; Rustin, P. Clinical presentations and laboratory investigations in respiratory chain deficiency. Eur. J. Pediatr. 1996, 155, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Esteitie, N.; Hinttala, R.; Wibom, R.; Nilsson, H.; Hance, N.; Naess, K.; Teär-Fahnehjelm, K.; Von Döbeln, U.; Majamaa, K.; Larsson, N.G. Secondary metabolic effects in complex I deficiency. Ann. Neurol. 2005, 58, 544–552. [Google Scholar] [CrossRef]
- Terburgh, K.; Lindeque, Z.; Mason, S.; van der Westhuizen, F.; Louw, R. Metabolomics of Ndufs4−/− skeletal muscle: Adaptive mechanisms converge at the ubiquinone-cycle. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 98–106. [Google Scholar] [CrossRef]
- Gielisch, I.; Meierhofer, D. Metabolome and proteome profiling of complex i deficiency induced by rotenone. J. Proteome Res. 2015, 14, 224–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrier Vergano, S.; Rao, M.; McCormack, S.; Ostrovsky, J.; Clarke, C.; Preston, J.; Bennett, M.J.; Yudkoff, M.; Xiao, R.; Falk, M.J. In vivo metabolic flux profiling with stable isotopes discriminates sites and quantifies effects of mitochondrial dysfunction in C. elegans. Mol. Genet. Metab. 2014, 111, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, P.G.; Higdon, R.; Kolker, N.; Bauman, A.T.; Ilkayeva, O.; Newgard, C.B.; Kolker, E.; Steele, L.M.; Sedensky, M.M. Comparison of proteomic and metabolomic profiles of mutants of the mitochondrial respiratory chain in Caenorhabditis elegans. Mitochondrion 2015, 20, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Garcia Canaveras, J.C.; Chen, Z.; Wang, L.; Liang, L.; Jang, C.; Mayr, J.A.; Zhang, Z.; Ghergurovich, J.M.; Zhan, L.; et al. Serine Catabolism Feeds NADH when Respiration Is Impaired. Cell Metab. 2020, 31, 809–821. [Google Scholar] [CrossRef]
- Naviaux, R.K. Oxidative shielding or oxidative stress? J. Pharmacol. Exp. Ther. 2012, 342, 608–618. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [Green Version]
- Ribas, G.S.; Vargas, C.R. Evidence that Oxidative Disbalance and Mitochondrial Dysfunction are Involved in the Pathophysiology of Fatty Acid Oxidation Disorders. Cell. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Violante, S.; Ijlst, L.; Ruiter, J.; Koster, J.; van Lenthe, H.; Duran, M.; de Almeida, I.T.; Wanders, R.J.A.; Houten, S.M.; Ventura, F.V. Substrate specificity of human carnitine acetyltransferase: Implications for fatty acid and branched-chain amino acid metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 773–779. [Google Scholar] [CrossRef] [Green Version]
- McCann, M.R.; De la Rosa, M.V.G.; Rosania, G.R.; Stringer, K.A. L-carnitine and acylcarnitines: Mitochondrial biomarkers for precision medicine. Metabolites 2021, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Xie, K.; Qin, Q.; Long, Z.; Yang, Y.; Peng, C.; Xi, C.; Li, L.; Wu, Z.; Daria, V.; Zhao, Y.; et al. High-Throughput Metabolomics for Discovering Potential Biomarkers and Identifying Metabolic Mechanisms in Aging and Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 602887. [Google Scholar] [CrossRef]
- Shao, Y.; Le, W. Recent advances and perspectives of metabolomics-based investigations in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Trillo, L.; Das, D.; Hsieh, W.; Medina, B.; Moghadam, S.; Lin, B.; Dang, V.; Sanchez, M.M.; De Miguel, Z.; Ashford, J.W.; et al. Ascending monoaminergic systems alterations in Alzheimer’s disease. Translating basic science into clinical care. Neurosci. Biobehav. Rev. 2013, 37, 1363–1379. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Holmes, C.; Sharabi, Y. Cerebrospinal fluid biomarkers of central catecholamine deficiency in Parkinson’s disease and other synucleinopathies. Brain 2012, 135, 1900–1913. [Google Scholar] [CrossRef]
- Martorana, A.; Koch, G. Is dopamine involved in Alzheimer’s disease? Front. Aging Neurosci. 2014, 6, 252. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, C.; Murphy, K.P.S.; Parent, M.; Levine, M.S. The Role of Dopamine in Huntington’s Disease, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 211. [Google Scholar]
- Xie, A.; Gao, J.; Xu, L.; Meng, D. Shared mechanisms of neurodegeneration in alzheimer’s disease and parkinson’s disease. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Nobili, A.; Latagliata, E.C.; Viscomi, M.T.; Cavallucci, V.; Cutuli, D.; Giacovazzo, G.; Krashia, P.; Rizzo, F.R.; Marino, R.; Federici, M.; et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Fridjonsdottir, E.; Shariatgorji, R.; Nilsson, A.; Vallianatou, T.; Odell, L.R.; Schembri, L.S.; Svenningsson, P.; Fernagut, P.O.; Crossman, A.R.; Bezard, E.; et al. Mass spectrometry imaging identifies abnormally elevated brain l-DOPA levels and extrastriatal monoaminergic dysregulation in l-DOPA-induced dyskinesia. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.; Niemelä, V.; Emami Khoonsari, P.; Sundblom, J.; Burman, J.; Landtblom, A.M.; Spjuth, O.; Nyholm, D.; Kultima, K. Alterations in the tyrosine and phenylalanine pathways revealed by biochemical profiling in cerebrospinal fluid of Huntington’s disease subjects. Sci. Rep. 2019, 9, 4129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schraml, E.; Quan, P.; Stelzer, I.; Fuchs, R.; Skalicky, M.; Viidik, A.; Schauenstein, K. Norepinephrine treatment and aging lead to systemic and intracellular oxidative stress in rats. Exp. Gerontol. 2007, 42, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.Y.; Roy, J.; Fung, M.L.; Heng, B.C.; Zhang, C.; Lim, L.W. Relationships between mitochondrial dysfunction and neurotransmission failure in Alzheimer’s disease. Aging Dis. 2020, 11, 1291–1316. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and the electron transport chain. Cell. Mol. Life Sci. 2017, 74, 3883–3896. [Google Scholar] [CrossRef] [PubMed]
- Shariatgorji, M.; Strittmatter, N.; Nilsson, A.; Källback, P.; Alvarsson, A.; Zhang, X.; Vallianatou, T.; Svenningsson, P.; Goodwin, R.J.A.; Andren, P.E. Simultaneous imaging of multiple neurotransmitters and neuroactive substances in the brain by desorption electrospray ionization mass spectrometry. Neuroimage 2016, 136, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.; Liu, L.; Meng, N.; Tang, Z.; Chua, K.; Chen, L.; Song, J. LC-MS-Based Urinary Metabolite Signatures in Idiopathic Parkinson’s Disease. J. Proteome Res. 2015, 14, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, Y.; Tsutsui, H.; Noda, T.; Akatsu, H.; Inoue, K. Widely targeted metabolomics of Alzheimer’s disease postmortem cerebrospinal fluid based on 9-fluorenylmethyl chloroformate derivatized ultra-high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2018, 1091, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, J.A.; Ammerman, G.M.; Reveles, J.M.; Ackermann, B.L. Analysis of glutamine, glutamate, pyroglutamate, and GABA in cerebrospinal fluid using ion pairing HPLC with positive electrospray LC/MS/MS. J. Neurosci. Methods 2008, 171, 190–196. [Google Scholar] [CrossRef]
- Go, Y.-M.; Uppal, K.; Walker, D.I.; Tran, V.; Dury, L.; Strobel, F.H.; Baubichon-Cortay, H.; Pennell, K.D.; Roede, J.R.; Jones, D.P. Mitochondrial metabolomics using high-resolution Fourier-transform mass spectrometry. In Mass Spectrometry in Metabolomics; Springer: Berlin/Heidelberg, Germany, 2014; pp. 43–73. [Google Scholar]
- Fishman, M.C. Sir Henry Hallett Dale and acetylcholine story. Yale J. Biol. Med. 1972, 45, 104–118. [Google Scholar]
- Rosas-Ballina, M.; Tracey, K.J. Cholinergic control of inflammation. J. Intern. Med. 2009, 265, 663–679. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Hughes, Z.A.; Kerns, E.H.; Lin, Q.; Beyer, C.E. Development of a liquid chromatography/tandem mass spectrometry method for the quantitation of acetylcholine and related neurotransmitters in brain microdialysis samples. J. Pharm. Biomed. Anal. 2007, 44, 586–593. [Google Scholar] [CrossRef]
- Peng, J.; Guo, K.; Xia, J.; Zhou, J.; Yang, J.; Westaway, D.; Wishart, D.S.; Li, L. Development of isotope labeling liquid chromatography mass spectrometry for mouse urine metabolomics: Quantitative metabolomic study of transgenic mice related to Alzheimer’s disease. J. Proteome Res. 2014, 13, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, C.; Simó, C.; Martín-Álvarez, P.J.; Kivipelto, M.; Winblad, B.; Cedazo-Mínguez, A.; Cifuentes, A. Toward a predictive model of Alzheimer’s disease progression using capillary electrophoresis-mass spectrometry metabolomics. Anal. Chem. 2012, 84, 8532–8540. [Google Scholar] [CrossRef]
- Walter, A.; Korth, U.; Hilgert, M.; Hartmann, J.; Weichel, O.; Hilgert, M.; Fassbender, K.; Schmitt, A.; Klein, J. Glycerophosphocholine is elevated in cerebrospinal fluid of Alzheimer patients. Neurobiol. Aging 2004, 25, 1299–1303. [Google Scholar] [CrossRef]
- Martinon, F. Mechanisms of uric acid crystal-mediated autoinflammation. Immunol. Rev. 2010, 233, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic signalling. Br. J. Pharmacol. 2006, 147, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic Signaling during Inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, S.; Gilchrist, R.B.; Richani, D. A sensitive method for the separation and quantification of low-level adenine nucleotides using porous graphitic carbon-based liquid chromatography and tandem mass spectrometry. J. Chromatogr. B 2017, 1061–1062, 445–451. [Google Scholar] [CrossRef]
- Esteve, C.; Jones, E.A.; Kell, D.B.; Boutin, H.; McDonnell, L.A. Mass spectrometry imaging shows major derangements in neurogranin and in purine metabolism in the triple-knockout 3×Tg Alzheimer mouse model. Biochim. Biophys. Acta Proteins Proteomics 2017, 1865, 747–754. [Google Scholar] [CrossRef]
- Lewitt, P.; Schultz, L.; Auinger, P.; Lu, M. CSF xanthine, homovanillic acid, and their ratio as biomarkers of Parkinson’s disease. Brain Res. 2011, 1408, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Trezzi, J.P.; Galozzi, S.; Jaeger, C.; Barkovits, K.; Brockmann, K.; Maetzler, W.; Berg, D.; Marcus, K.; Betsou, F.; Hiller, K.; et al. Distinct metabolomic signature in cerebrospinal fluid in early parkinson’s disease. Mov. Disord. 2017, 32, 1401–1408. [Google Scholar] [CrossRef]
- De Lau, L.M.L.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M.B. Serum uric acid levels and the risk of Parkinson disease. Ann. Neurol. 2005, 58, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; LeWitt, P.A.; Watts, A.; Kieburtz, K.; Rudolph, A.; Schwid, S.R.; PSG DATA TOP invertigators. CSF as well as serum urate are predictors of Parkinson’s disease progression. In Proceedings of the 10th International Conference of Parkinson’s Disease and Movement Disorders, Kyoto, Japan, 28 October–2 November 2006. [Google Scholar]
- Younes-Mhenni, S.; Frih-Ayed, M.; Kerkeni, A.; Bost, M.; Chazot, G. Peripheral blood markers of oxidative stress in Parkinson’s disease. Eur. Neurol. 2007, 58, 78–83. [Google Scholar] [CrossRef]
- Graham, S.F.; Kumar, P.; Bahado-Singh, R.O.; Robinson, A.; Mann, D.; Green, B.D. Novel metabolite biomarkers of Huntington’s disease as detected by high-resolution mass spectrometry. J. Proteome Res. 2016, 15, 1592–1601. [Google Scholar] [CrossRef]
- Gmitterova, K.; Heinemann, U.; Gawinecka, J.; Varges, D.; Ciesielczyk, B.; Valkovic, P.; Benetin, J.; Zerr, I. 8-OHdG in cerebrospinal fluid as a marker of oxidative stress in various neurodegenerative diseases. Neurodegener. Dis. 2009, 6, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Stoessel, D.; Schulte, C.; Teixeira dos Santos, M.C.; Scheller, D.; Rebollo-Mesa, I.; Deuschle, C.; Walther, D.; Schauer, N.; Berg, D.; Nogueira da Costa, A.; et al. Promising metabolite profiles in the plasma and CSF of early clinical Parkinson’s disease. Front. Aging Neurosci. 2018, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Lei, S.; Levytskyy, R.; Pappa, A.; Panayiotidis, M.I.; Cerny, R.L.; Khalimonchuk, O.; Powers, R.; Franco, R. Glucose Metabolism and AMPK Signaling Regulate Dopaminergic Cell Death Induced by Gene (α-Synuclein)-Environment (Paraquat) Interactions. Mol. Neurobiol. 2017, 54, 3825–3842. [Google Scholar] [CrossRef]
- Lei, S.; Zavala-Flores, L.; Garcia-Garcia, A.; Nandakumar, R.; Huang, Y.; Madayiputhiya, N.; Stanton, R.C.; Dodds, E.D.; Powers, R.; Franco, R. Alterations in energy/redox metabolism induced by mitochondrial and environmental toxins: A specific role for glucose-6-phosphate-dehydrogenase and the pentose phosphate pathway in paraquat toxicity. ACS Chem. Biol. 2014, 9, 2032–2048. [Google Scholar] [CrossRef] [PubMed]
- Hämäläinen, R.H.; Manninen, T.; Koivumäki, H.; Kislin, M.; Otonkoski, T.; Suomalainen, A. Tissue-and cell-type–specific manifestations of heteroplasmic mtDNA 3243A> G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl. Acad. Sci. USA 2013, 110, E3622–E3630. [Google Scholar] [CrossRef] [Green Version]
- Teslaa, T.; Teitell, M.A. Pluripotent stem cell energy metabolism: An update. EMBO J. 2015, 34, 138–153. [Google Scholar] [CrossRef]
- Yokota, M.; Hatakeyama, H.; Okabe, S.; Ono, Y.; Goto, Y. Mitochondrial respiratory dysfunction caused by a heteroplasmic mitochondrial DNA mutation blocks cellular reprogramming. Hum. Mol. Genet. 2015, 24, 4698–4709. [Google Scholar] [CrossRef] [Green Version]
- Iannetti, E.F.; Prigione, A.; Smeitink, J.A.M.; Koopman, W.J.H.; Beyrath, J.; Renkema, H. Live-imaging readouts and cell models for phenotypic profiling of mitochondrial function. Front. Genet. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, M.; Hatakeyama, H.; Ono, Y.; Kanazawa, M.; Goto, Y. Mitochondrial respiratory dysfunction disturbs neuronal and cardiac lineage commitment of human iPSCs. Cell Death Dis. 2018, 8, e2551. [Google Scholar] [CrossRef]
- Okarmus, J.; Bogetofte, H.; Schmidt, S.I.; Ryding, M.; García-López, S.; Ryan, B.J.; Martínez-Serrano, A.; Hyttel, P.; Meyer, M. Lysosomal perturbations in human dopaminergic neurons derived from induced pluripotent stem cells with PARK2 mutation. Sci. Rep. 2020, 10, 10278. [Google Scholar] [CrossRef]
- Okarmus, J.; Havelund, J.F.; Ryding, M.; Schmidt, S.I.; Bogetofte, H.; Færgeman, N.J.; Hyttel, P.; Meyer, M. Identification of bioactive metabolites in human iPSC-derived dopaminergic neurons with PARK2 mutation: Altered mitochondrial and energy metabolism. bioRxiv 2020. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Suleiman, J.; Almannai, M.; Scaglia, F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol. Genet. Metab. 2018, 125, 315–321. [Google Scholar] [CrossRef]
- Amore, G.; Romagnoli, M.; Carbonelli, M.; Barboni, P.; Carelli, V.; La Morgia, C. Therapeutic Options in Hereditary Optic Neuropathies. Drugs 2020, 81, 57–86. [Google Scholar] [CrossRef]
- Bu, X.L.; Rao, P.P.N.; Wang, Y.J. Anti-amyloid Aggregation Activity of Natural Compounds: Implications for Alzheimer’s Drug Discovery. Mol. Neurobiol. 2016, 53, 3565–3575. [Google Scholar] [CrossRef] [PubMed]
- Titov, D.V.; Cracan, V.; Goodman, R.P.; Peng, J.; Grabarek, Z.; Mootha, V.K. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 2016, 352, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivelä, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428.e5. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.R.; Ong, S.E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F.; et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 2016, 5, e10575. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of neuronal energy deficiency by mtor inhibition as a treatment for mitochondria-related neurodegeneration. Elife 2016, 5, e13378. [Google Scholar] [CrossRef] [PubMed]


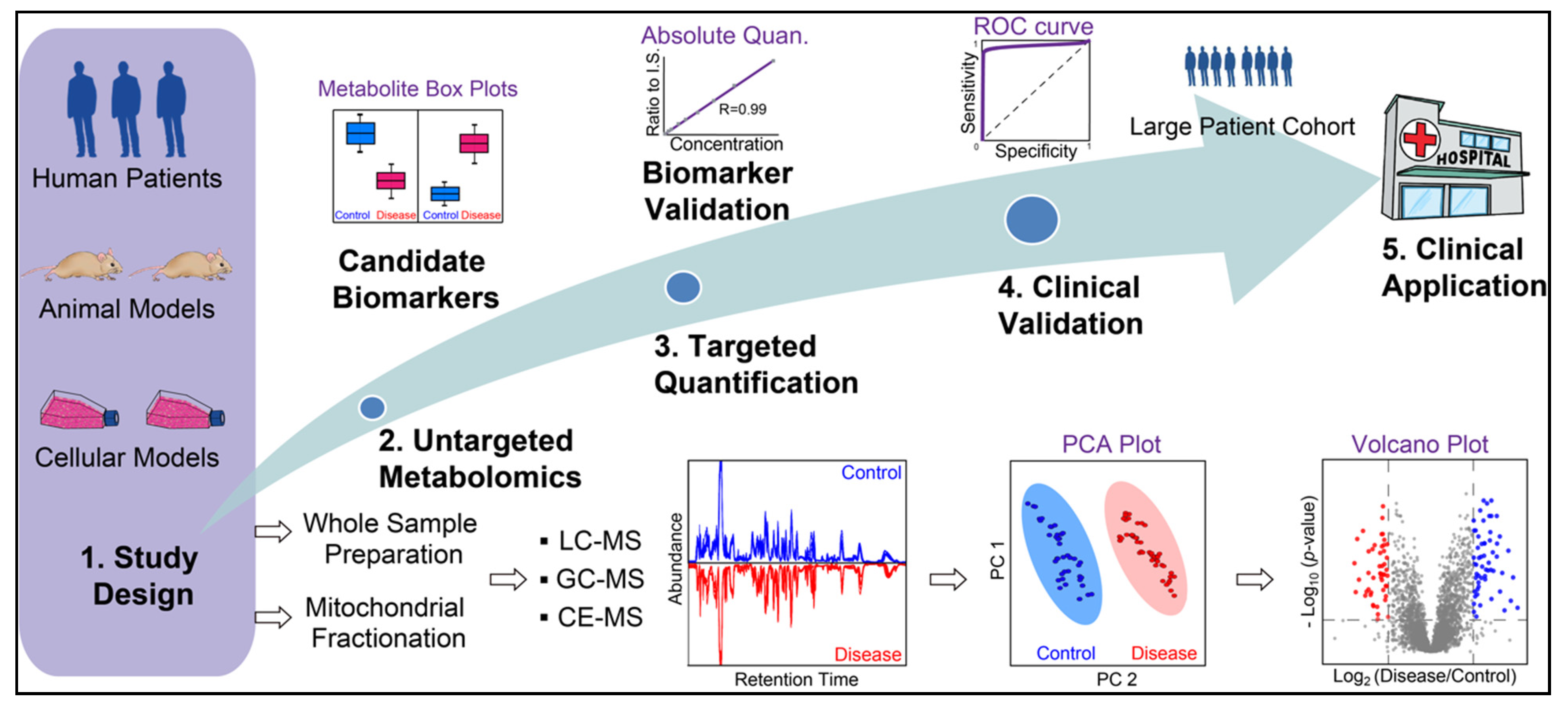

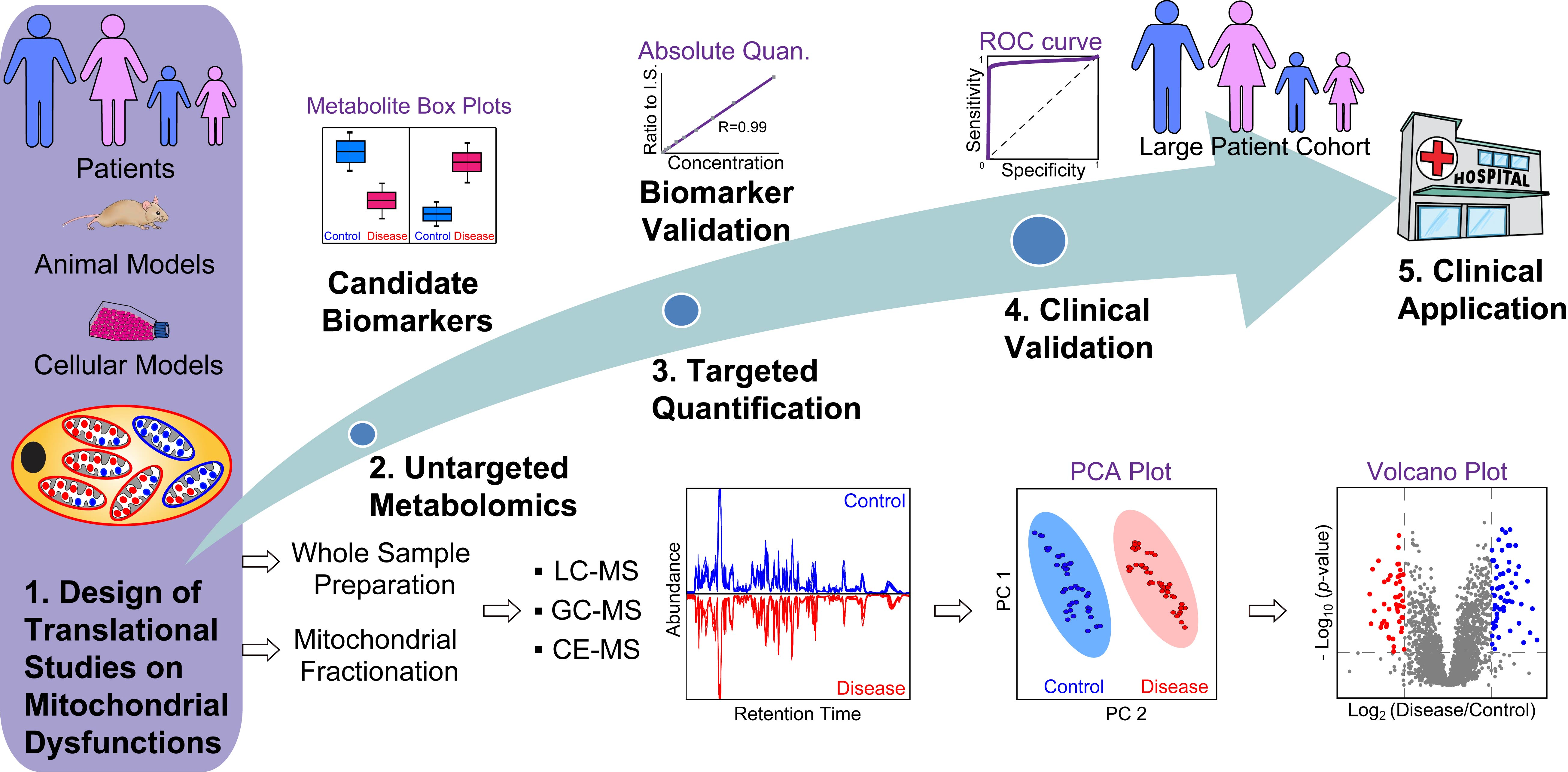{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Pattern of Inheritance | Variants | Onset | Key Clinical Features |
|---|---|---|---|---|
| KSS | Mt | Deletion of 4977 bp of mtDNA | Late childhood | Progressive ophthalmoplegia, pigmentary retinopathy, cerebellar ataxia, cardiac conduction defects |
| Leigh | Mt or nuclear (AR) | m.10191T > C (ND3) m.10197G > A (ND3) m.13573G > A (ND5) m.14487T > C (ND6) SURF1 COX10 COX15 SCO2 | Infancy or early childhood | Ataxia, intellectual retardation, hypotonia, motor delay, cardiomyopathy, brainstem dysfunction, and demyelination |
| LHON | Mt | m.11778G > A (ND4) m.3460G > A (ND1) m.14484T > C (ND6) | Early adulthood | Optic neuropathy with acute or subacute loss of central vision |
| MELAS | Mt | m.3243A > G (mt-tRNALeu(UUR)) m.1630A > G (mt-tRNAVal) m.3697G > A (ND1) m.13514A > G (ND5) m.14453G > A (ND6) | Childhood or early adulthood | Encephalopathy, lactic acidosis, stroke-like episodes, myopathy, seizures, cognitive deficit, recurrent migraines, gastrointestinal dysmotility |
| MILS | Mt | m.8993T > G (ATPase6) m.4681C > T (ND2) m.135513G > A (ND5) | Infancy or early childhood | Encephalopathy, developmental delay, hypotonia, lactic acidosis, seizures, ataxia, optic atrophy, dysphagia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Uittenbogaard, M.; Hao, L.; Chiaramello, A. Clinical Insights into Mitochondrial Neurodevelopmental and Neurodegenerative Disorders: Their Biosignatures from Mass Spectrometry-Based Metabolomics. Metabolites 2021, 11, 233. https://doi.org/10.3390/metabo11040233
Li H, Uittenbogaard M, Hao L, Chiaramello A. Clinical Insights into Mitochondrial Neurodevelopmental and Neurodegenerative Disorders: Their Biosignatures from Mass Spectrometry-Based Metabolomics. Metabolites. 2021; 11(4):233. https://doi.org/10.3390/metabo11040233
Chicago/Turabian StyleLi, Haorong, Martine Uittenbogaard, Ling Hao, and Anne Chiaramello. 2021. "Clinical Insights into Mitochondrial Neurodevelopmental and Neurodegenerative Disorders: Their Biosignatures from Mass Spectrometry-Based Metabolomics" Metabolites 11, no. 4: 233. https://doi.org/10.3390/metabo11040233
APA StyleLi, H., Uittenbogaard, M., Hao, L., & Chiaramello, A. (2021). Clinical Insights into Mitochondrial Neurodevelopmental and Neurodegenerative Disorders: Their Biosignatures from Mass Spectrometry-Based Metabolomics. Metabolites, 11(4), 233. https://doi.org/10.3390/metabo11040233