
Reconstruction of a Genome-Scale Metabolic Model of Streptomyces albus J1074: Improved Engineering Strategies in Natural Product Synthesis

 , , , and
, , , and 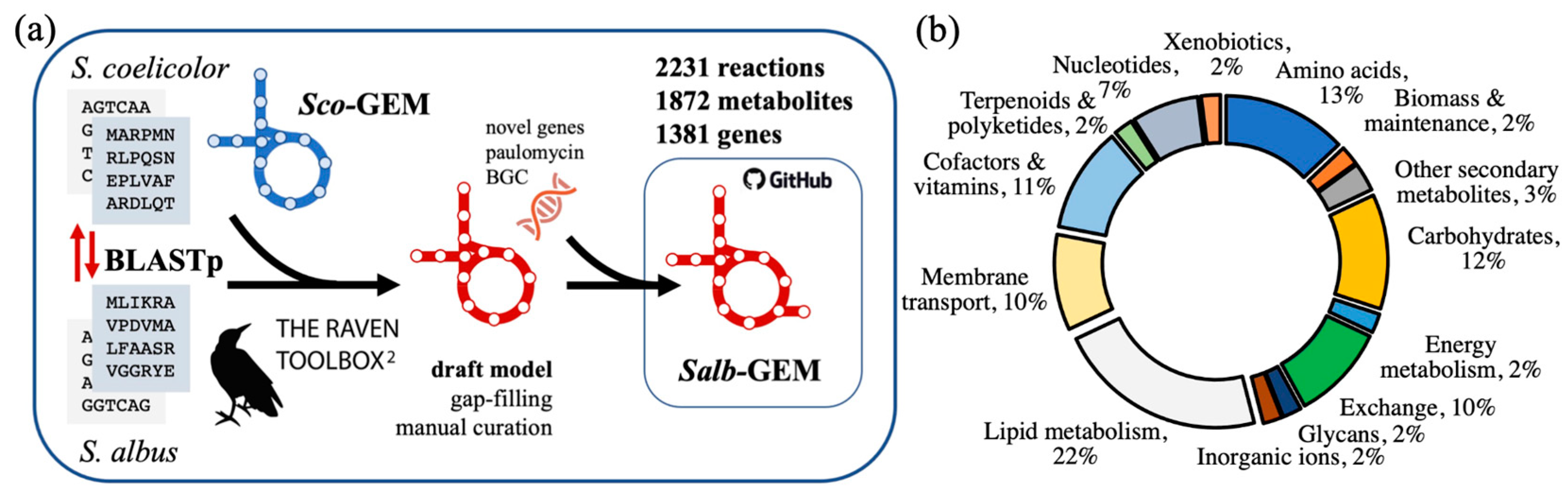{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Reconstruction of Draft Genome-Scale Model
2.2. Further Curation towards Salb-GEM
2.3. Calibrating Energetic Parameters from Cultivations
2.4. Functional Comparison to Sco-GEM Reference Model
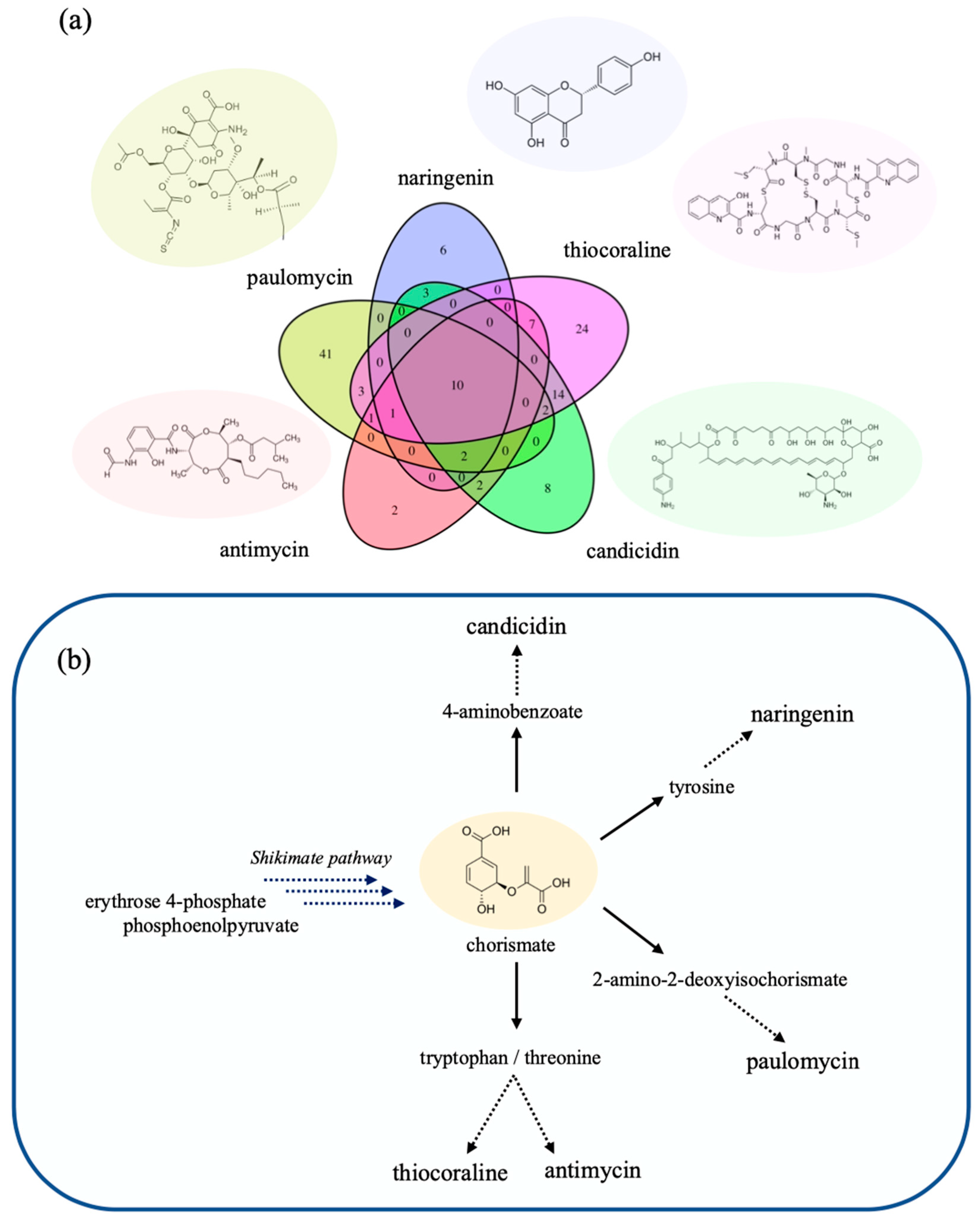
2.5. Design Strategies for Improved Compound Production in S. albus
3. Materials and Methods
3.1. Model Reconstruction
3.1.1. Homology-Based Reconstruction
3.1.2. Definition of Biomass Composition and Energetic Parameters
3.1.3. Gap-Filling
3.1.4. Curation of Reaction Reversibility
3.1.5. Model Distribution
3.2. Model Analysis
3.3. Cultivation and Data Collection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hopwood, D.A. Forty years of genetics with Streptomyces: From in vivo through in vitro to in silico. Microbiology 1999, 145, 2183–2202. [Google Scholar] [CrossRef] [PubMed]
- Palazzotto, E.; Tong, Y.; Lee, S.Y.; Weber, T. Synthetic biology and metabolic engineering of actinomycetes for natural product discovery. Biotechnol. Adv. 2019, 37, 107366. [Google Scholar] [CrossRef]
- Kim, J.H.; Komatsu, M.; Shin-ya, K.; Omura, S.; Ikeda, H. Distribution and functional analysis of the phosphopantetheinyl transferase superfamily in Actinomycetales microorganisms. Proc. Natl. Acad. Sci. USA 2018, 115, 6828–6833. [Google Scholar] [CrossRef] [PubMed]
- Kallifidas, D.; Jiang, G.; Ding, Y.; Luesch, H. Rational engineering of Streptomyces albus J1074 for the overexpression of secondary metabolite gene clusters. Microb. Cell Fact. 2018, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Myronovskyi, M.; Tokovenko, B.; Brötz, E.; Rückert, C.; Kalinowski, J.; Luzhetskyy, A. Genome rearrangements of Streptomyces albus J1074 lead to the carotenoid gene cluster activation. Appl. Microbiol. Biotechnol. 2014, 98, 795–806. [Google Scholar] [CrossRef]
- Liu, X.; Liu, D.; Xu, M.; Tao, M.; Bai, L.; Deng, Z.; Pfeifer, B.A.; Jiang, M. Reconstitution of Kinamycin Biosynthesis within the Heterologous Host Streptomyces albus J1074. J. Nat. Prod. 2018, 81, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Zaburannyi, N.; Rabyk, M.; Ostash, B.; Fedorenko, V.; Luzhetskyy, A. Insights into naturally minimised Streptomyces albus J1074 genome. BMC Genomics 2014, 15, 97. [Google Scholar] [CrossRef]
- Chater, A.N.K.F.; Lynn, D. Streptomyces albus G Mutants Defective in the SaZGI Res tric tion-Modifica tion S ys tern. J. Gen. Microbiol. 1980, 116, 323–334. [Google Scholar]
- Lombó, F.; Velasco, A.; Castro, A.; de la Calle, F.; Braña, A.F.; Sánchez-Puelles, J.M.; Méndez, C.; Salas, J. A Deciphering the biosynthesis pathway of the antitumor thiocoraline from a marine actinomycete and its expression in two streptomyces species. Chembiochem 2006, 7, 366–376. [Google Scholar] [CrossRef]
- Myronovskyi, M.; Rosenkränzer, B.; Nadmid, S.; Pujic, P.; Normand, P.; Luzhetskyy, A. Generation of a cluster-free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab. Eng. 2018, 49, 316–324. [Google Scholar] [CrossRef]
- Ahmed, Y.; Rebets, Y.; Tokovenko, B.; Brötz, E.; Luzhetskyy, A. Identification of butenolide regulatory system controlling secondary metabolism in Streptomyces albus J1074. Sci. Rep. 2017, 7, 9784. [Google Scholar] [CrossRef]
- Bilyk, B.; Horbal, L.; Luzhetskyy, A. Chromosomal position effect influences the heterologous expression of genes and biosynthetic gene clusters in Streptomyces albus J1074. Microb. Cell Fact. 2017, 16, 5. [Google Scholar] [CrossRef]
- Li, J.; Xie, Z.; Wang, M.; Ai, G.; Chen, Y. Identification and analysis of the paulomycin biosynthetic gene cluster and titer improvement of the paulomycins in Streptomyces paulus NRRL 8115. PLoS ONE 2015, 10, e0120542. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Rodríguez, M.; Braña, A.F.; Méndez, C.; Salas, J.A.; Olano, C. New insights into paulomycin biosynthesis pathway in Streptomyces albus J1074 and generation of novel derivatives by combinatorial biosynthesis. Microb. Cell Fact. 2016, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Merlo, M.E.; Hodgson, D.A.; Wellington, E.M.H.; Takano, E.; Breitling, R. Metabolic modeling and analysis of the metabolic switch in Streptomyces coelicolor. BMC Genom. 2010, 11, 202. [Google Scholar] [CrossRef]
- Fernández- De la Hoz, J.; Méndez, C.; Salas, J.A.; Olano, C. Novel Bioactive Paulomycin Derivatives Produced by Streptomyces albus J1074. Molecules 2017, 22, 1758. [Google Scholar]
- Koshla, O.; Yushchuk, O.; Ostash, I.; Dacyuk, Y.; Myronovskyi, M.; Jäger, G.; Süssmuth, R.D.; Luzhetskyy, A.; Byström, A.; Kirsebom, L.A.; et al. Gene miaA for post-transcriptional modification of tRNAXXA is important for morphological and metabolic differentiation in Streptomyces. Mol. Microbiol. 2019, 112, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Koshla, O.T.; Rokytskyy, I.V.; Ostash, I.S.; Busche, T.; Kalinowski, J.; Mösker, E.; Süssmuth, R.D.; Fedorenko, V.O.; Ostash, B.O. Secondary Metabolome and Transcriptome of Streptomyces albus J1074 in Liquid Medium SG2. Cytol. Genet. 2019, 53, 1–7. [Google Scholar] [CrossRef]
- Marín, L.; Gutiérrez-Del-Río, I.; Yagüe, P.; Manteca, Á.; Villar, C.J.; Lombó, F. De Novo Biosynthesis of Apigenin, Luteolin, and Eriodictyol in the Actinomycete Streptomyces albus and Production Improvement by Feeding and Spore Conditioning. Front. Microbiol. 2017, 8, 921. [Google Scholar] [CrossRef] [PubMed]
- Marín, L.; Gutiérrez-del-Río, I.; Entrialgo-Cadierno, R.; Villar, C.J.; Lombó, F. De novo biosynthesis of myricetin, kaempferol and quercetin in Streptomyces albus and Streptomyces coelicolor. PLoS ONE 2018, 13, e0207278. [Google Scholar] [CrossRef]
- O’Brien, E.J.; Monk, J.M.; Palsson, B.O. Using Genome-Scale Models to Predict Biological Capabilities. Cell 2015, 161, 971–987. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef]
- Sulheim, S.; Kumelj, T.; Van Dissel, D.; Salehzadeh-Yazdi, A.; Du, C.; Nieselt, K.; Almaas, E.; Wentzel, A.; Kerkhoven, E. Genome-scale model constrained by proteomics reveals metabolic rearrangements in the heterologous host streptomyces coelicolor. bioRxiv 2019. [Google Scholar] [CrossRef]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef]
- Agren, R.; Liu, L.; Shoaie, S.; Vongsangnak, W.; Nookaew, I.; Nielsen, J. The RAVEN Toolbox and Its Use for Generating a Genome-scale Metabolic Model for Penicillium chrysogenum. PLoS Comput. Biol. 2013, 9, e1002980. [Google Scholar] [CrossRef]
- Alam, M.T.; Medema, M.H.; Takano, E.; Breitling, R. Comparative genome-scale metabolic modeling of actinomycetes: The topology of essential core metabolism. FEBS Lett. 2011, 585, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.; Chater, K.; Cerdeño-Tárraga, A.-M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Olano, C.; García, I.; González, A.; Rodriguez, M.; Rozas, D.; Rubio, J.; Sánchez-Hidalgo, M.; Braña, A.F.; Méndez, C.; Salas, J.A. Activation and identification of five clusters for secondary metabolites in Streptomyces albus J1074. Microb. Biotechnol. 2014, 7, 242–256. [Google Scholar] [CrossRef]
- Kim, J.N.; Kim, Y.; Jeong, Y.; Roe, J.H.; Kim, B.G.; Cho, B.K. Comparative genomics reveals the core and accessory genomes of streptomyces speciesj. J. Microbiol. Biotechnol. 2015, 25, 1599–1605. [Google Scholar] [CrossRef] [PubMed]
- Seipke, R.F. Strain-level diversity of secondary metabolism in Streptomyces albus. PLoS ONE 2015, 10, e0116457. [Google Scholar]
- Chen, Y.; Nielsen, J. Energy metabolism controls phenotypes by protein efficiency and allocation. Proc. Natl. Acad. Sci. USA 2019, 116, 17592–17597. [Google Scholar] [CrossRef]
- Klappenbach, J.A.; Dunbar, J.M.; Schmidt, T.M. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 2000, 66, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Lee, S.Y.; Kim, T.Y.; Woo, H.M. In silico identification of gene amplification targets for improvement of lycopene production. Appl. Environ. Microbiol. 2010, 76, 3097–3105. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Álvarez, R.; Botas, A.; Albillos, S.M.; Rumbero, A.; Martín, J.F.; Liras, P. Molecular genetics of naringenin biosynthesis, a typical plant secondary metabolite produced by Streptomyces clavuligerus. Microb. Cell Fact. 2015, 14, 178. [Google Scholar] [CrossRef]
- Arabolaza, A.; Shillito, M.E.; Lin, T.-W.; Diacovich, L.; Melgar, M.; Pham, H.; Amick, D.; Gramajo, H.; Tsai, S.-C. Crystal structures and mutational analyses of acyl-CoA carboxylase beta subunit of Streptomyces coelicolor. Biochemistry 2010, 49, 7367–7376. [Google Scholar] [CrossRef]
- Rodríguez, E.; Banchio, C.; Diacovich, L.; Bibb, M.J.; Gramajo, H. Role of an essential acyl coenzyme A carboxylase in the primary and secondary metabolism of Streptomyces coelicolor A3(2). Appl. Environ. Microbiol. 2001, 67, 4166–4176. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Yu, O. Metabolic engineering of flavonoids in plants and microorganisms. Appl. Microbiol. Biotechnol. 2011, 91, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Dunstan, M.S.; Robinson, C.J.; Jervis, A.J.; Yan, C.; Carbonell, P.; Hollywood, K.A.; Currin, A.; Swainston, N.; Le Feuvre, R.; Micklefield, J.; et al. Engineering Escherichia coli towards de novo production of gatekeeper (2S)-flavanones: Naringenin, pinocembrin, eriodictyol and homoeriodictyol. Synth. Biol. 2020, 5, ysaa012. [Google Scholar] [CrossRef] [PubMed]
- Noor, E.; Haraldsdóttir, H.S.; Milo, R.; Fleming, R.M.T. Consistent estimation of Gibbs energy using component contribution. PLoS Comput. Biol. 2014, 9, e1003098. [Google Scholar] [CrossRef] [PubMed]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Burton, K. A study of the conditions and mechanism of the diphenylamine reaction for the colorimetric estimation of deoxyribonucleic acid. Biochem. J. 1956, 62, 315–323. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kittikunapong, C.; Ye, S.; Magadán-Corpas, P.; Pérez-Valero, Á.; Villar, C.J.; Lombó, F.; Kerkhoven, E.J. Reconstruction of a Genome-Scale Metabolic Model of Streptomyces albus J1074: Improved Engineering Strategies in Natural Product Synthesis. Metabolites 2021, 11, 304. https://doi.org/10.3390/metabo11050304
Kittikunapong C, Ye S, Magadán-Corpas P, Pérez-Valero Á, Villar CJ, Lombó F, Kerkhoven EJ. Reconstruction of a Genome-Scale Metabolic Model of Streptomyces albus J1074: Improved Engineering Strategies in Natural Product Synthesis. Metabolites. 2021; 11(5):304. https://doi.org/10.3390/metabo11050304
Chicago/Turabian StyleKittikunapong, Cheewin, Suhui Ye, Patricia Magadán-Corpas, Álvaro Pérez-Valero, Claudio J. Villar, Felipe Lombó, and Eduard J. Kerkhoven. 2021. "Reconstruction of a Genome-Scale Metabolic Model of Streptomyces albus J1074: Improved Engineering Strategies in Natural Product Synthesis" Metabolites 11, no. 5: 304. https://doi.org/10.3390/metabo11050304
APA StyleKittikunapong, C., Ye, S., Magadán-Corpas, P., Pérez-Valero, Á., Villar, C. J., Lombó, F., & Kerkhoven, E. J. (2021). Reconstruction of a Genome-Scale Metabolic Model of Streptomyces albus J1074: Improved Engineering Strategies in Natural Product Synthesis. Metabolites, 11(5), 304. https://doi.org/10.3390/metabo11050304