
Metabolic Response of Pancreatic Carcinoma Cells under Treatment with Dichloroacetate

 ,
, 
Abstract
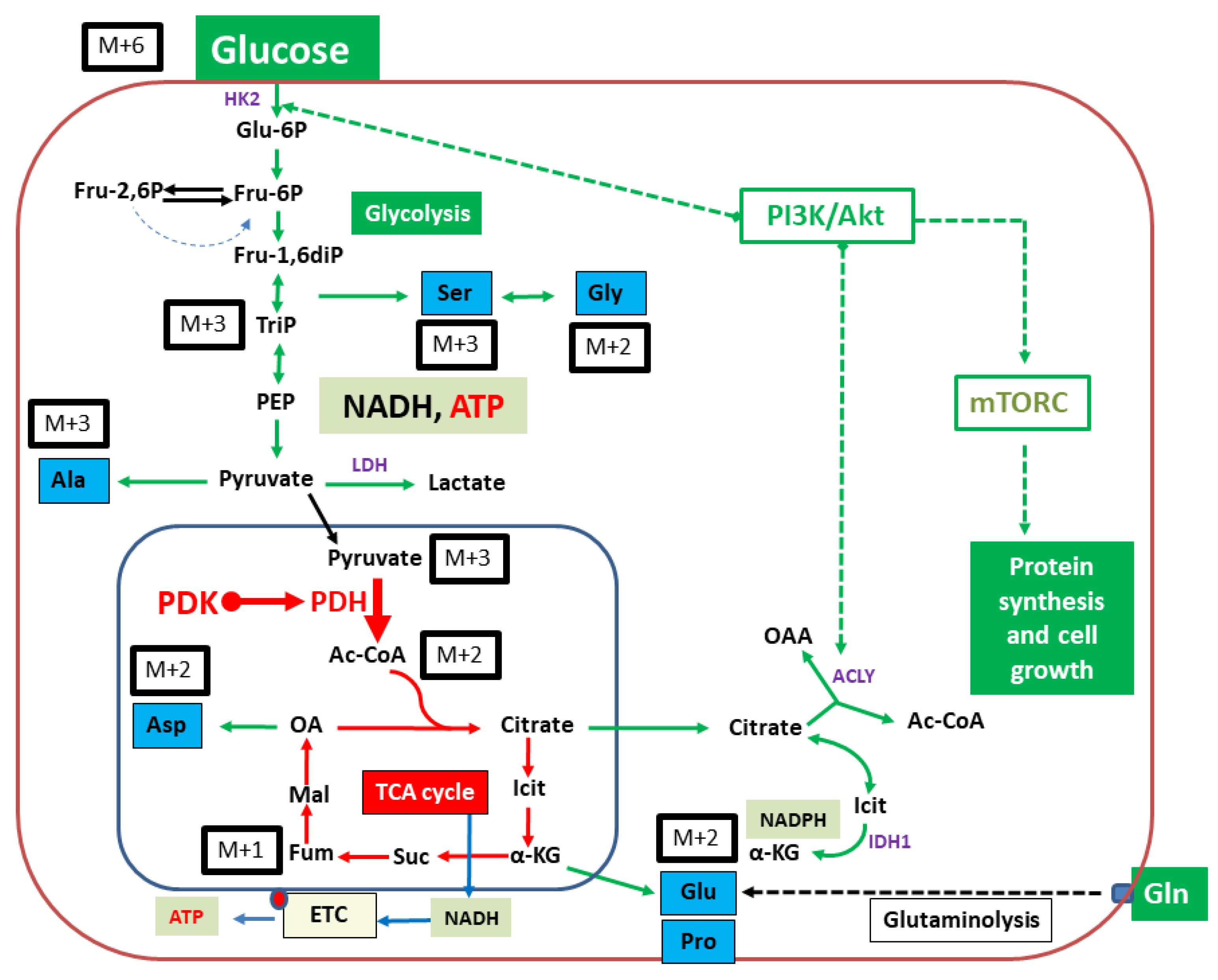
:1. Introduction
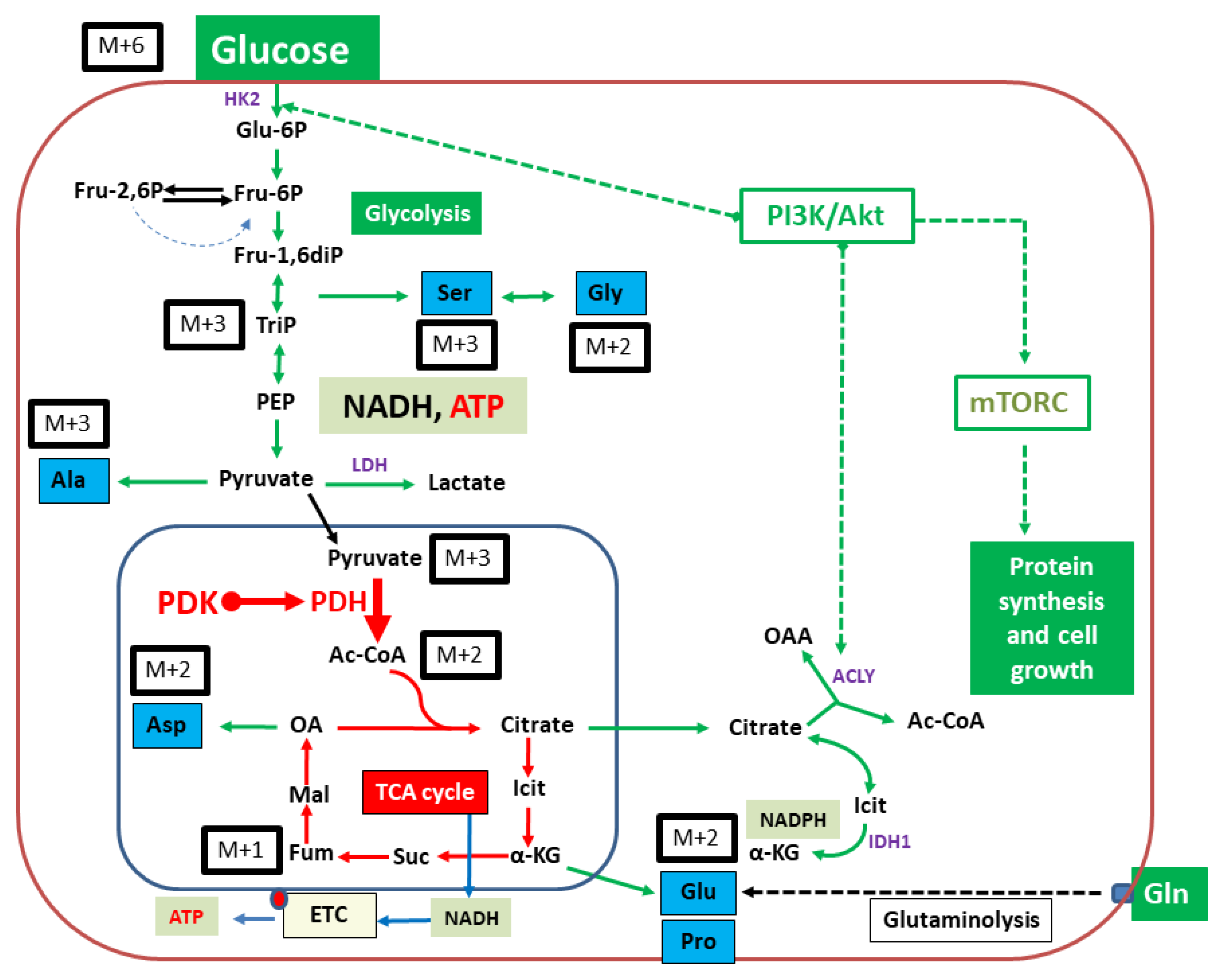
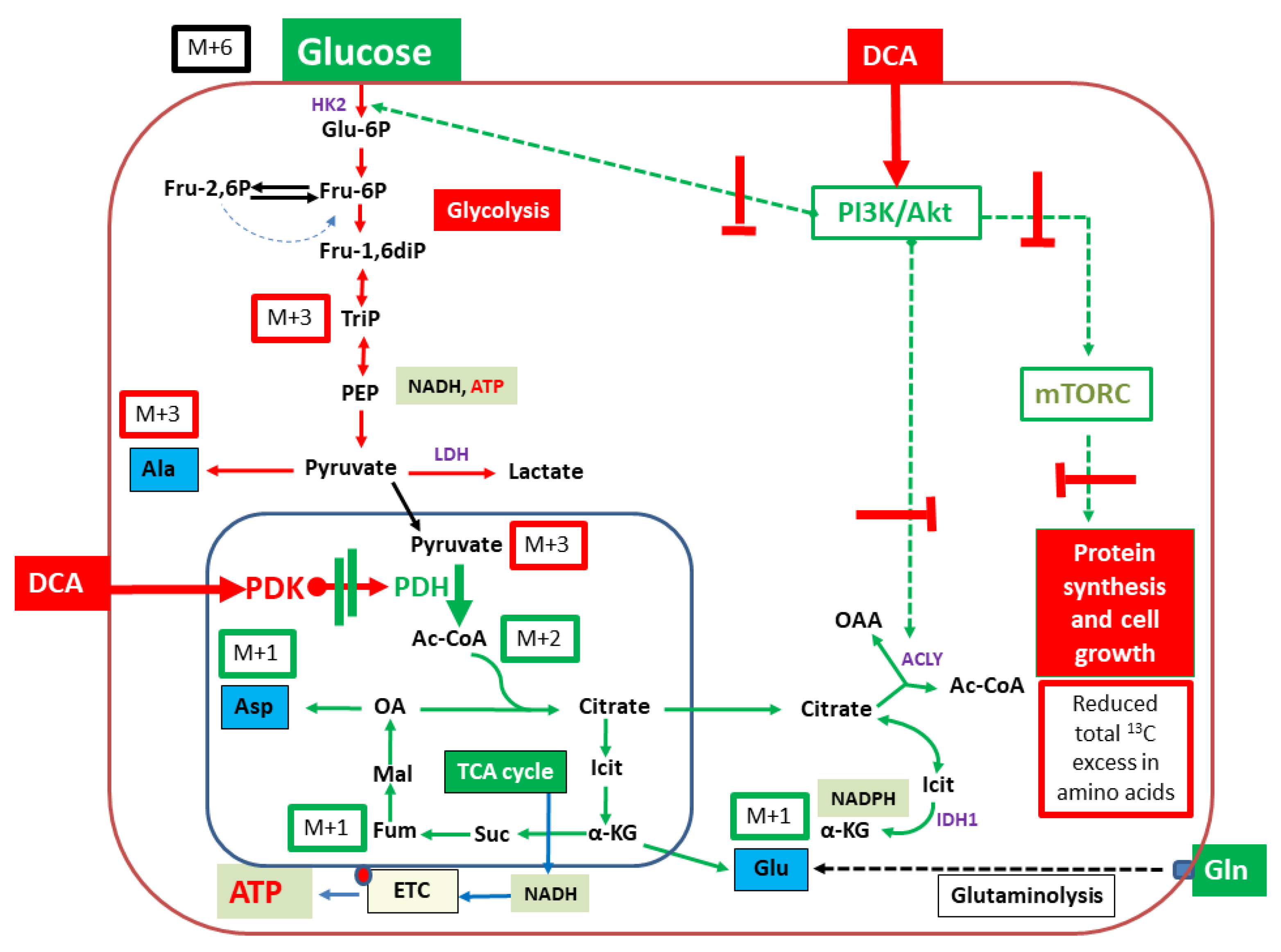
2. Results
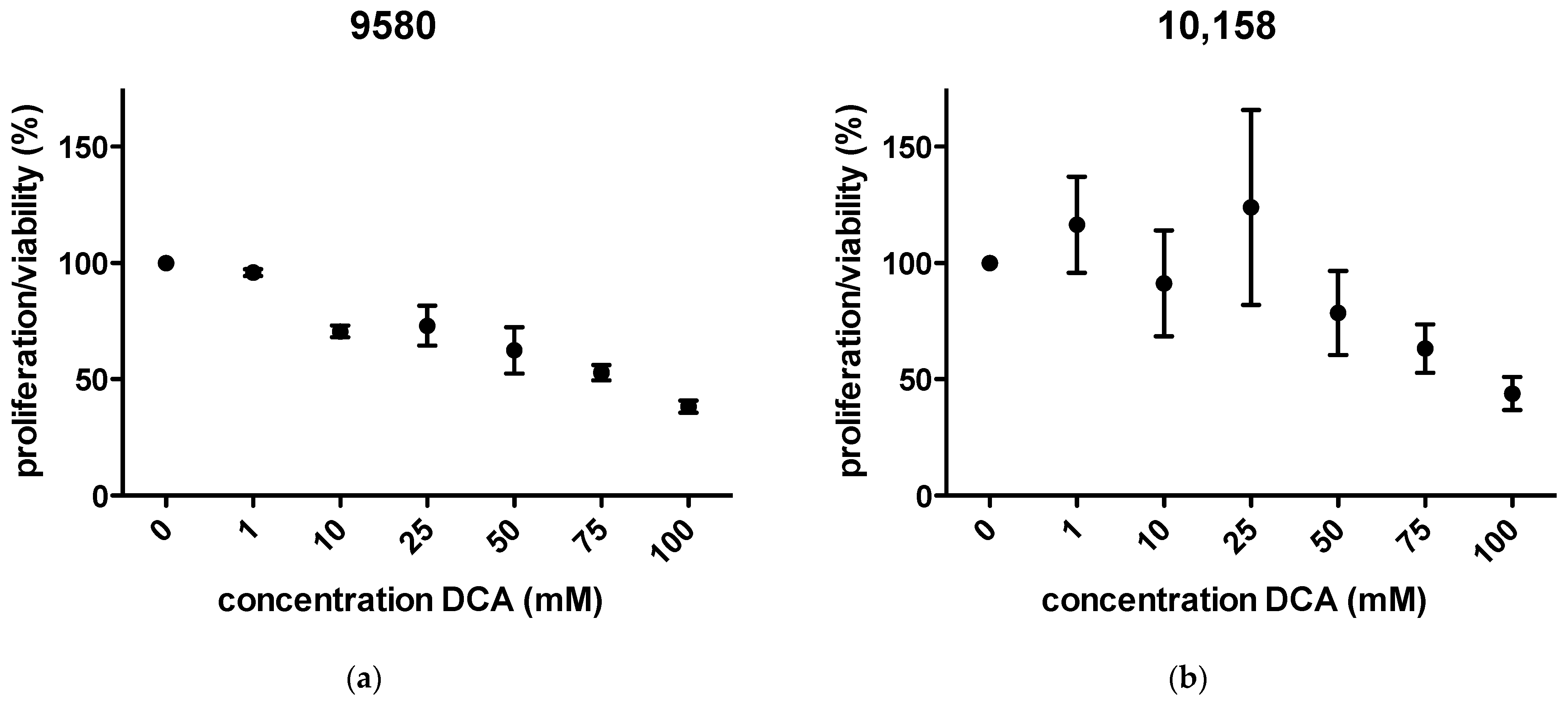
2.1. Proliferation/Viability
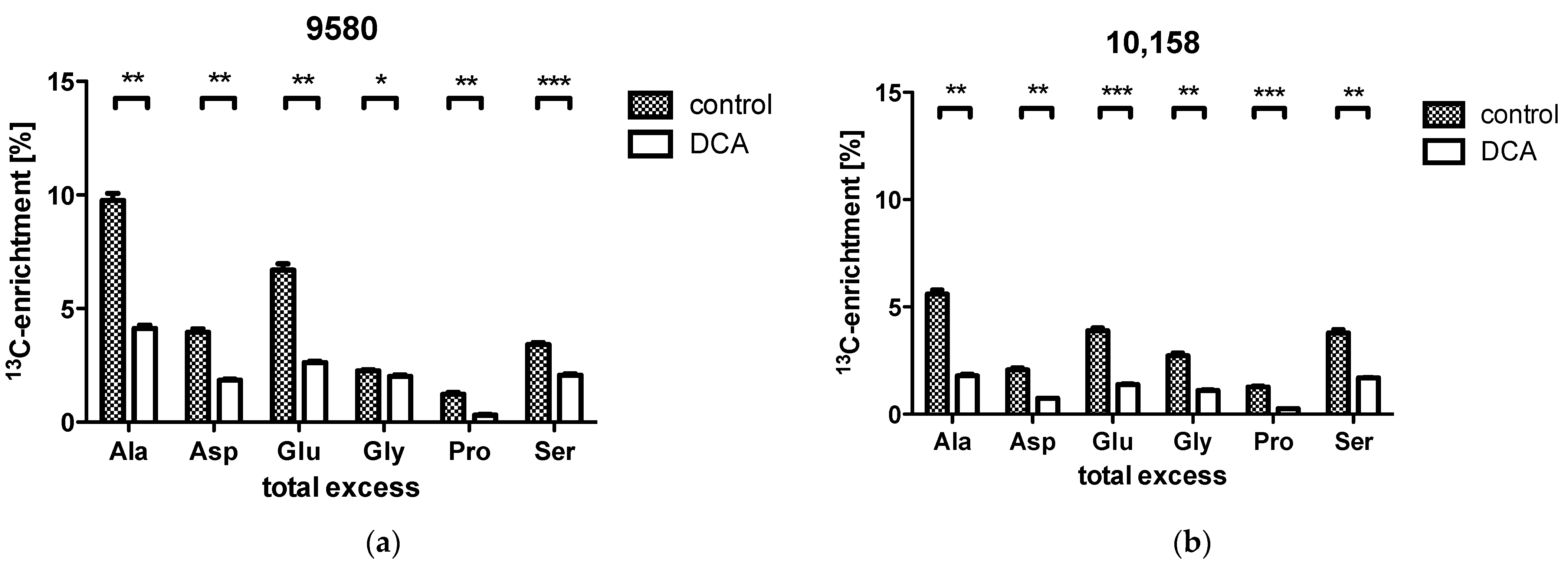
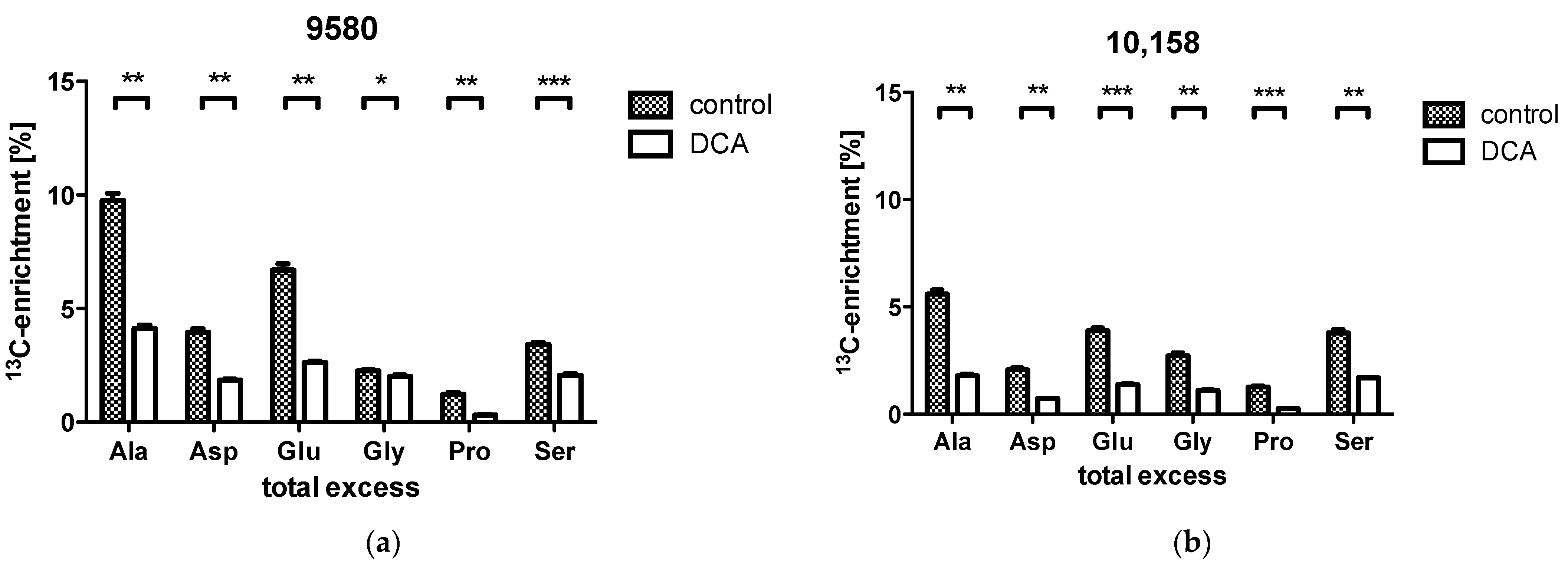
2.2. Isotopologue Distribution and 13C-Enrichment in Selected Amino Acids
2.3. Overall 13C-Contents of Amino Acids
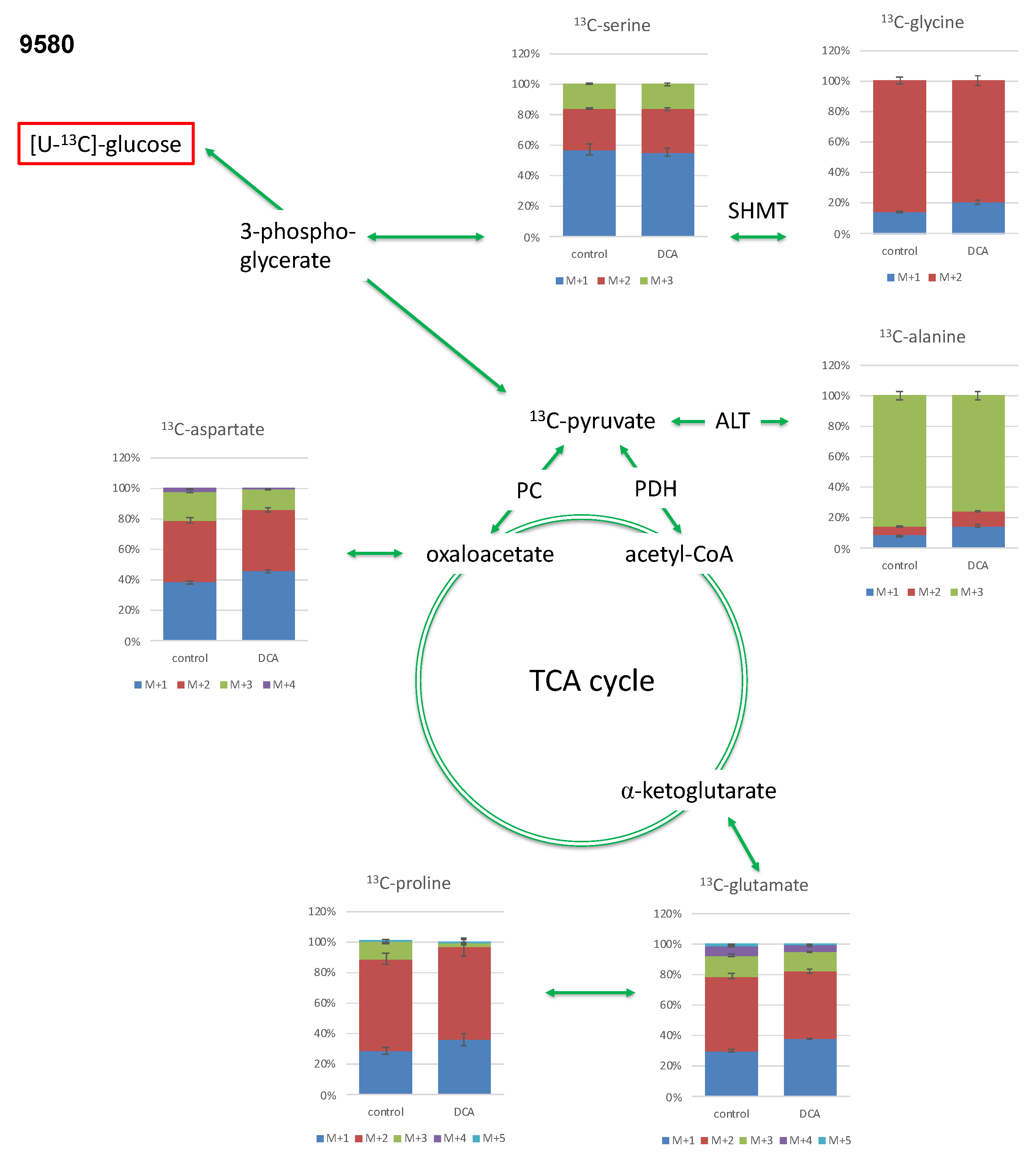
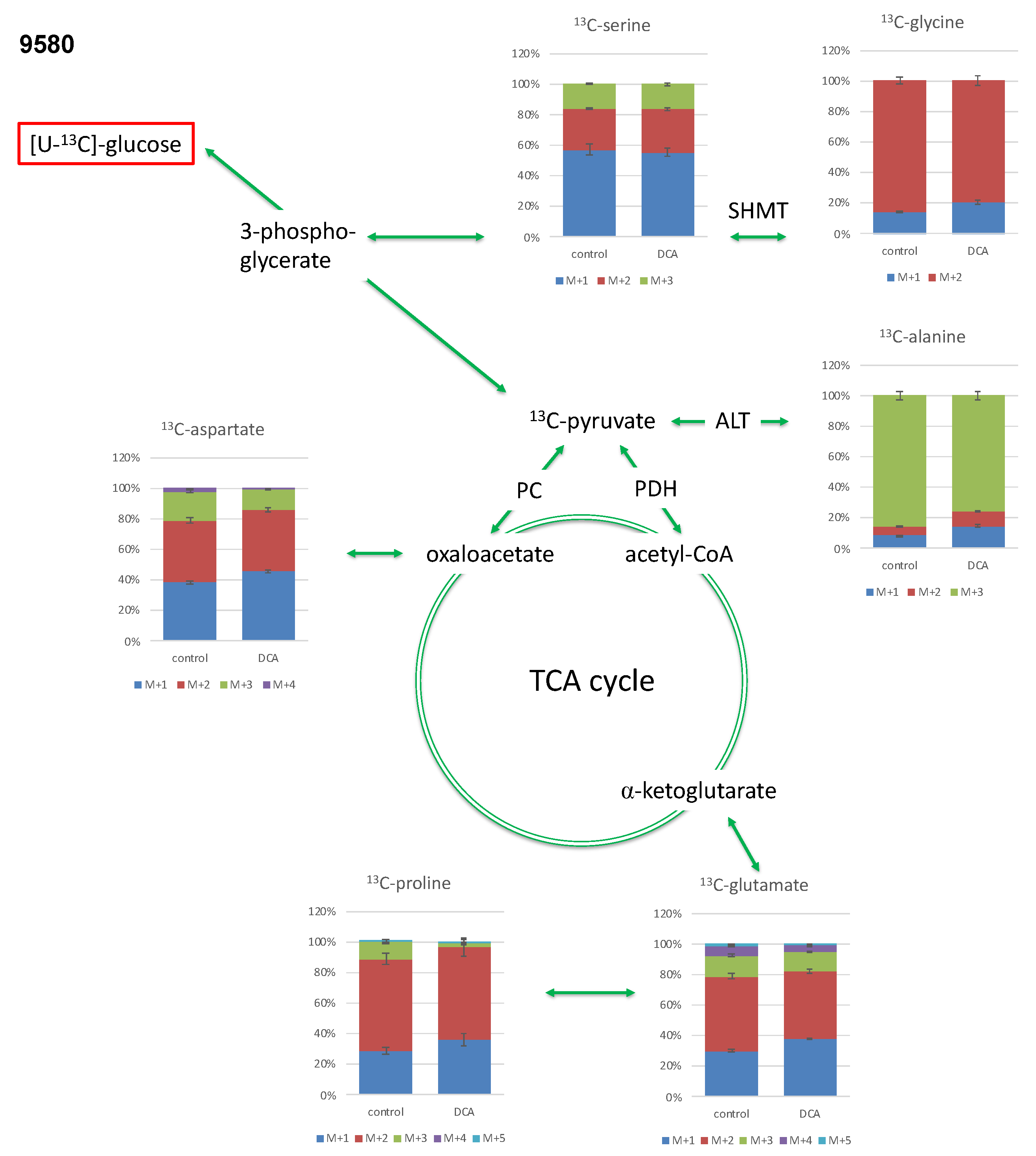
2.3.1. Isotopologue Profiles of Amino Acids from 9580 Cells
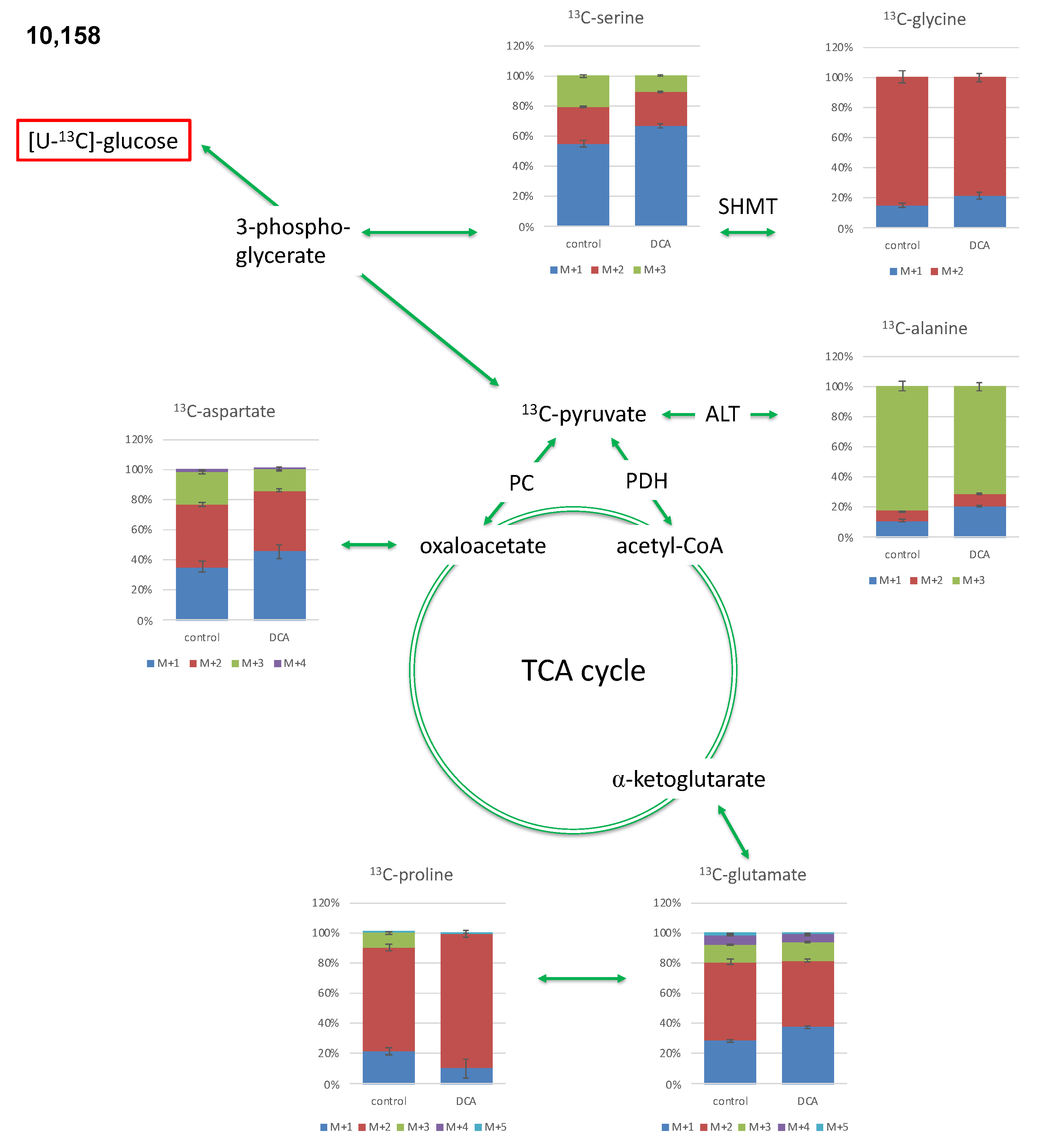
2.3.2. Isotopologue Profiles of Amino Acids from 10,158 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. WST-1 Cell Viability/Proliferation Assay
4.3. Treatment of Cells with DCA and Incubation with [U-13C6]Glucose
4.4. Isotopologue Profiling of Cell Lines Exposed to DCA
4.5. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA A Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [Green Version]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef]
- Aichler, M.; Seiler, C.; Tost, M.; Siveke, J.; Mazur, P.K.; Da Silva-Buttkus, P.; Bartsch, D.K.; Langer, P.; Chiblak, S.; Dürr, A.; et al. Origin of pancreatic ductal adenocarcinoma from atypical flat lesions: A comparative study in transgenic mice and human tissues. J. Pathol. 2011, 226, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology 2012, 142, 730–733.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.A.; Brisset, J.-C.; Zhang, Y.; Bednar, F.; Pierre, J.; Heist, K.A.; Galbán, C.J.; Galbán, S.; Di Magliano, M.P. Metastatic Pancreatic Cancer Is Dependent on Oncogenic Kras in Mice. PLoS ONE 2012, 7, e49707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Britten, C.D. PI3K and MEK inhibitor combinations: Examining the evidence in selected tumor types. Cancer Chemother. Pharmacol. 2013, 71, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic Modulation of Glioblastoma with Dichloroacetate. Sci. Transl. Med. 2010, 2, 31–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Zhu, D.; Zhu, L.; Hou, Y.; Hou, L.; Huang, X.; Li, L.; Wang, Y.; Li, L.; Zou, H.; et al. Dichloroacetate Overcomes Oxaliplatin Chemoresistance in Colorectal Cancer through the miR-543/PTEN/Akt/mTOR Pathway. J. Cancer 2019, 10, 6037–6047. [Google Scholar] [CrossRef] [PubMed]
- Kolesnik, D.; Pyaskovskaya, O.; Yakshibaeva, Y.; Solyanik, G. Time-dependent cytotoxicity of dichloroacetate and metformin against Lewis lung carcinoma. Exp. Oncol. 2019, 41, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Lam, Y.-M.; Leung, Y.-C.; Hu, X.; Chen, X.; Cheung, E.; Tam, K.Y. Combined use of arginase and dichloroacetate exhibits anti-proliferative effects in triple negative breast cancer cells. J. Pharm. Pharmacol. 2019, 71, 306–315. [Google Scholar] [CrossRef]
- Li, B.; Li, X.; Xiong, H.; Zhou, P.; Ni, Z.; Yang, T.; Zhang, Y.; Zeng, Y.; He, J.; Yang, F.; et al. Inhibition of COX2 enhances the chemosensitivity of dichloroacetate in cervical cancer cells. Oncotarget 2017, 8, 51748–51757. [Google Scholar] [CrossRef] [Green Version]
- Abildgaard, C.; Dahl, C.; Abdul-Al, A.; Christensen, A.; Guldberg, P. Inhibition of retinoic acid receptor beta signaling confers glycolytic dependence and sensitization to dichloroacetate in melanoma cells. Oncotarget 2017, 8, 84210–84223. [Google Scholar] [CrossRef] [Green Version]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Stacpoole, P.W. The pharmacology of dichloroacetate. Metabolism 1989, 38, 1124–1144. [Google Scholar] [CrossRef]
- James, M.O.; Yan, Z.; Cornett, R.; Jayanti, V.M.; Henderson, G.N.; Davydova, N.; Katovich, M.J.; Pollock, B.; Stacpoole, P.W. Pharmacokinetics and metabolism of [14C]dichloroacetate in male Sprague-Dawley rats. Identification of glycine conjugates, including hippurate, as urinary metabolites of dichloroacetate. Drug Metab. Dispos. 1998, 26, 1134–1143. [Google Scholar]
- Yan, Z.; Henderson, G.N.; James, M.O.; Stacpoole, P.W. Determination of dichloroacetate and its metabolites in human plasma by gas chromatography–mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1997, 703, 75–84. [Google Scholar] [CrossRef]
- Shroads, A.L.; Coats, B.S.; Langaee, T.; Shuster, J.J.; Stacpoole, P.W. Chloral hydrate, through biotransformation to dichloroacetate, inhibits maleylacetoacetate isomerase and tyrosine catabolism in humans. Drug Metab. Pers. Ther. 2015, 30, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Gkiouli, M.; Biechl, P.; Eisenreich, W.; Otto, A. Otto Diverse Roads Taken by 13C-Glucose-Derived Metabolites in Breast Cancer Cells Exposed to Limiting Glucose and Glutamine Conditions. Cells 2019, 8, 1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerecker, B.; Biechl, P.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; Schwaiger, M.; Eisenreich, W. Diverse metabolic response of cancer cells treated with a 213Bi-anti-EGFR-immunoconjugate. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Otto, A.M. Metabolic Constants and Plasticity of Cancer Cells in a Limiting Glucose and Glutamine Microenvironment—A Pyruvate Perspective. Front. Oncol. 2020, 10, 596197. [Google Scholar] [CrossRef]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.W.M.; Lane, A.N.; Higashi, R.M.; A Farag, M.; Gao, H.; Bousamra, M.; Miller, D.M. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Mol. Cancer 2009, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.H.; Dickens, F.; Kaiser-Wilhelm-Institut for Biology. The Metabolism of Tumours; Investigations from the Kaiser Wilhelm Institute for Biology, Berlin-Dahlem; Constable & Co. Ltd.: London, UK, 1930. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta Bioenerg. 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Liu, L.; Chai, W.; Zhao, T.; Jin, X.; Guo, X.; Han, L.; Yuan, C. Dichloroacetic acid upregulates apoptosis of ovarian cancer cells by regulating mitochondrial function. OncoTargets Ther. 2019, 12, 1729–1739. [Google Scholar] [CrossRef] [Green Version]
- Hur, H.; Xuan, Y.; Kim, Y.B.; Lee, G.; Shim, W.; Yun, J.; Ham, I.-H.; Han, S.-U. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int. J. Oncol. 2012, 42, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerecker, B.; Seidl, C.; Pirsig, S.; Bruchelt, G.; Senekowitsch-Schmidtke, R. DCA promotes progression of neuroblastoma tumors in nude mice. Am. J. Cancer Res. 2015, 5, 812–820. [Google Scholar] [PubMed]
- Feuerecker, B.; Pirsig, S.; Seidl, C.; Aichler, M.; Feuchtinger, A.; Bruchelt, G.; Senekowitsch-Schmidtke, R. Lipoic acid inhibits cell proliferation of tumor cells in vitro and in vivo. Cancer Biol. Ther. 2012, 13, 1425–1435. [Google Scholar] [CrossRef] [Green Version]
- Morfouace, M.; Lalier, L.; Bahut, M.; Bonnamain, V.; Naveilhan, P.; Guette, C.; Oliver, L.; Gueguen, N.; Reynier, P.; Vallette, F.M. Comparison of Spheroids Formed by Rat Glioma Stem Cells and Neural Stem Cells Reveals Differences in Glucose Metabolism and Promising Therapeutic Applications*. J. Biol. Chem. 2012, 287, 33664–33674. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.; Coomber, B.L. Pyruvate dehydrogenase kinase expression and metabolic changes following dichloroacetate exposure in anoxic human colorectal cancer cells. Exp. Cell Res. 2015, 331, 73–81. [Google Scholar] [CrossRef]
- Stockwin, L.H.; Yu, S.X.; Borgel, S.; Hancock, C.; Wolfe, T.L.; Phillips, L.R.; Hollingshead, M.G.; Newton, D.L. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int. J. Cancer 2010, 127, 2510–2519. [Google Scholar] [CrossRef]
- Gong, F.; Peng, X.; Sang, Y.; Qiu, M.; Luo, C.; He, Z.; Zhao, X.; Tong, A. Dichloroacetate induces protective autophagy in LoVo cells: Involvement of cathepsin D/thioredoxin-like protein 1 and Akt-mTOR-mediated signaling. Cell Death Dis. 2013, 4, e913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhu, Y.; Sun, Q.; Yu, C.; Chen, L.; Tian, Y.; Yan, J. Reversal of the Warburg effect with DCA in PDGFtreated human PASMC is potentiated by pyruvate dehydrogenase kinase1 inhibition mediated through blocking Akt/GSK3beta signalling. Int. J. Mol. Med. 2018, 42, 1391–1400. [Google Scholar] [PubMed]
- Sradhanjali, S.; Tripathy, D.; Rath, S.; Mittal, R.; Reddy, M.M. Overexpression of pyruvate dehydrogenase kinase 1 in retinoblastoma: A potential therapeutic opportunity for targeting vitreous seeds and hypoxic regions. PLoS ONE 2017, 12, e0177744. [Google Scholar] [CrossRef]
- Misbin, R.I. Effects of dichloroacetate on lipid metabolism in isolated rat liver cells. Diabetes 1979, 28, 265–271. [Google Scholar] [CrossRef]
- Diehl, F.F.; Lewis, C.A.; Fiske, B.P.; Heiden, M.G.V. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 2019, 1, 861–867. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Eakin, R.; Morgan, L.; Gregg, C.; Matwiyoff, N. Carbon-13 nuclear magnetic resonance spectroscopy of living cells and their metabolism of a specifically labeled13C substrate. FEBS Lett. 1972, 28, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Serrao, E.M.; Brindle, K.M. Dynamic nuclear polarisation: The future of imaging in oncology? Porto Biomed. J. 2017, 2, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Feuerecker, B.; Durst, M.; Michalik, M.; Schneider, G.; Saur, D.; Menzel, M.; Schwaiger, M.; Schilling, F. Hyperpolarized 13C Diffusion MRS of Co-Polarized Pyruvate and Fumarate to Measure Lactate Export and Necrosis. J. Cancer 2017, 8, 3078–3085. [Google Scholar] [CrossRef] [Green Version]
- Feuerecker, B.; Michalik, M.; Hundshammer, C.; Schwaiger, M.; Bruchertseifer, F.; Morgenstern, A.; Seidl, C. Assessment of 213Bi-anti-EGFR MAb treatment efficacy in malignant cancer cells with [1-13C]pyruvate and [18F]FDG. Sci. Rep. 2019, 9, 8294. [Google Scholar] [CrossRef] [Green Version]
- Bliemsrieder, E.; Kaissis, G.; Grashei, M.; Topping, G.; Altomonte, J.; Hundshammer, C.; Lohöfer, F.; Heid, I.; Keim, D.; Gebrekidan, S.; et al. Hyperpolarized 13C pyruvate magnetic resonance spectroscopy for in vivo metabolic phenotyping of rat HCC. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Hundshammer, C.; Braeuer, M.; Müller, C.A.; Hansen, A.E.; Schillmaier, M.; Düwel, S.; Feuerecker, B.; Glaser, S.J.; Haase, A.; Weichert, W.; et al. Simultaneous characterization of tumor cellularity and the Warburg effect with PET, MRI and hyperpolarized 13C-MRSI. Theranostics 2018, 8, 4765–4780. [Google Scholar] [CrossRef]
- Mishkovsky, M.; Anderson, B.; Karlsson, M.; Lerche, M.H.; Sherry, A.D.; Gruetter, R.; Kovacs, Z.; Comment, A. Measuring glucose cerebral metabolism in the healthy mouse using hyperpolarized 13C magnetic resonance. Sci. Rep. 2017, 7, 11719. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Suh, E.H.; Sharma, G.; Khemtong, C.; Sherry, A.D.; Kovacs, Z. Probing carbohydrate metabolism using hyperpolarized 13 C-labeled molecules. NMR Biomed. 2019, 32, e4018. [Google Scholar] [CrossRef]
- Brender, J.R.; Kishimoto, S.; Merkle, H.; Reed, G.; Hurd, R.E.; Chen, A.P.; Ardenkjaer-Larsen, J.H.; Munasinghe, J.; Saito, K.; Seki, T.; et al. Dynamic Imaging of Glucose and Lactate Metabolism by (13)C-MRS without Hyperpolarization. Sci Rep. 2019, 9, 3410. [Google Scholar] [CrossRef] [PubMed]
- Eylert, E.; Herrmann, V.; Jules, M.; Gillmaier, N.; Lautner, M.; Buchrieser, C.; Eisenreich, W.; Heuner, K. Isotopologue profiling of Legionella pneumophila: Role of serine and glucose as carbon substrates. J. Biol. Chem. 2010, 285, 22232–22243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eylert, E.; Schar, J.; Mertins, S.; Stoll, R.; Bacher, A.; Goebel, W.; Eisenreich, W. Carbon metabolism of Listeria monocytogenes growing inside macrophages. Mol. Microbiol. 2008, 69, 1008–1017. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 9580 | 10,158 | |||
| Control | DCA | Control | DCA | |
| Alanine | ||||
| M + 1 | 0.90% ± 0.03% | 0.70% ± 0.02% ** | 0.68% ± 0.07% | 0.45% ± 0.01% * |
| M + 2 | 0.65% ± 0.02% | 0.44% ± 0.02% *** | 0.40% ± 0.03% | 0.18% ± 0.01% ** |
| M + 3 | 9.02% ± 0.31% | 3.60% ± 0.13% ** | 5.10% ± 0.20% | 1.53% ± 0.06% ** |
| Aspartate | ||||
| M + 1 | 3.28% ± 0.11% | 1.99% ± 0.05% * | 1.54% ± 0.16% | 0.80% ± 0.08% * |
| M + 2 | 3.47% ± 0.13% | 1.76% ± 0.06% ** | 1.83% ± 0.06% | 0.71% ± 0.02% ** |
| M + 3 | 1.63% ± 0.06% | 0.59% ± 0.03% *** | 0.93% ± 0.05% | 0.24% ± 0.03% *** |
| M + 4 | 0.18% ± 0.01% | 0.03% ± 0.00% ** | 0.07% ± 0.01% | 0.00% ± 0.00% ** |
| Glutamate | ||||
| M + 1 | 5.03% ± 0.14% | 2.67% ± 0.05% ** | 2.76% ± 0.09% | 1.38% ± 0.03% ** |
| M + 2 | 8.12% ± 0.30% | 3.14% ± 0.10% ** | 5.09% ± 0.15% | 1.62% ± 0.04% *** |
| M + 3 | 2.29% ± 0.11% | 0.84% ± 0.03% ** | 1.15% ± 0.05% | 0.45% ± 0.03% ** |
| M + 4 | 1.02% ± 0.07% | 0.32% ± 0.01% ** | 0.61% ± 0.03% | 0.18% ± 0.01% ** |
| M + 5 | 0.24% ± 0.02% | 0.06% ± 0.00% ** | 0.13% ± 0.00% | 0.04% ± 0.00% *** |
| Glycine | ||||
| M + 1 | 0.35% ± 0.02% | 0.47% ± 0.03% * | 0.44% ± 0.04% | 0.27% ± 0.03% ** |
| M + 2 | 2.08% ± 0.06% | 1.77% ± 0.07% * | 2.49% ± 0.12% | 0.97% ± 0.04% ** |
| Proline | ||||
| M + 1 | 0.95% ± 0.07% | 0.33% ± 0.04% *** | 0.73% ± 0.08% | 0.07% ± 0.04% ** |
| M + 2 | 2.01% ± 0.12% | 0.56% ± 0.05% ** | 2.31% ± 0.07% | 0.57% ± 0.02% *** |
| M + 3 | 0.37% ± 0.04% | 0.02% ± 0.02% ** | 0.33% ± 0.02% | 0.00% ± 0.00% ** |
| M + 4 | 0.00% ± 0.00% | 0.00% ± 0.00% | 0.00% ± 0.00% | 0.00% ± 0.00% |
| M + 5 | 0.00% ± 0.00% | 0.00% ± 0.00% | 0.00% ± 0.00% | 0.00% ± 0.00% |
| Serine | ||||
| M + 1 | 3.67% ± 0.23% | 2.13% ± 0.10% ** | 3.80% ± 0.15% | 2.34% ± 0.04% ** |
| M + 2 | 1.75% ± 0.04% | 1.09% ± 0.04% ** | 1.68% ± 0.05% | 0.80% ± 0.02% ** |
| M + 3 | 1.02% ± 0.03% | 0.63% ± 0.04% ** | 1.41% ± 0.08% | 0.38% ± 0.02% ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feuerecker, B.; Biechl, P.; Veltkamp, C.; Saur, D.; Eisenreich, W. Metabolic Response of Pancreatic Carcinoma Cells under Treatment with Dichloroacetate. Metabolites 2021, 11, 350. https://doi.org/10.3390/metabo11060350
Feuerecker B, Biechl P, Veltkamp C, Saur D, Eisenreich W. Metabolic Response of Pancreatic Carcinoma Cells under Treatment with Dichloroacetate. Metabolites. 2021; 11(6):350. https://doi.org/10.3390/metabo11060350
Chicago/Turabian StyleFeuerecker, Benedikt, Philipp Biechl, Christian Veltkamp, Dieter Saur, and Wolfgang Eisenreich. 2021. "Metabolic Response of Pancreatic Carcinoma Cells under Treatment with Dichloroacetate" Metabolites 11, no. 6: 350. https://doi.org/10.3390/metabo11060350
APA StyleFeuerecker, B., Biechl, P., Veltkamp, C., Saur, D., & Eisenreich, W. (2021). Metabolic Response of Pancreatic Carcinoma Cells under Treatment with Dichloroacetate. Metabolites, 11(6), 350. https://doi.org/10.3390/metabo11060350