
Sigma-1 Receptor Promotes Mitochondrial Bioenergetics by Orchestrating ER Ca2+ Leak during Early ER Stress

 , , , , ,
, , , , ,  and
and 
Abstract
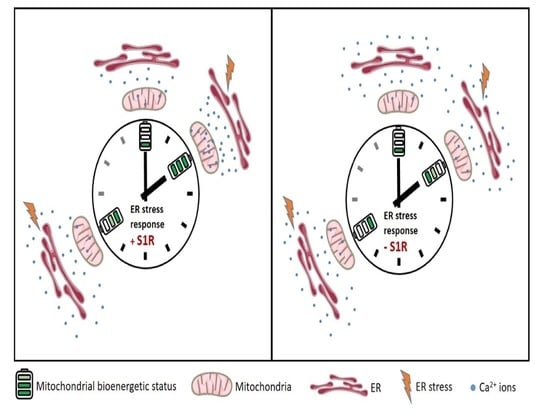
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
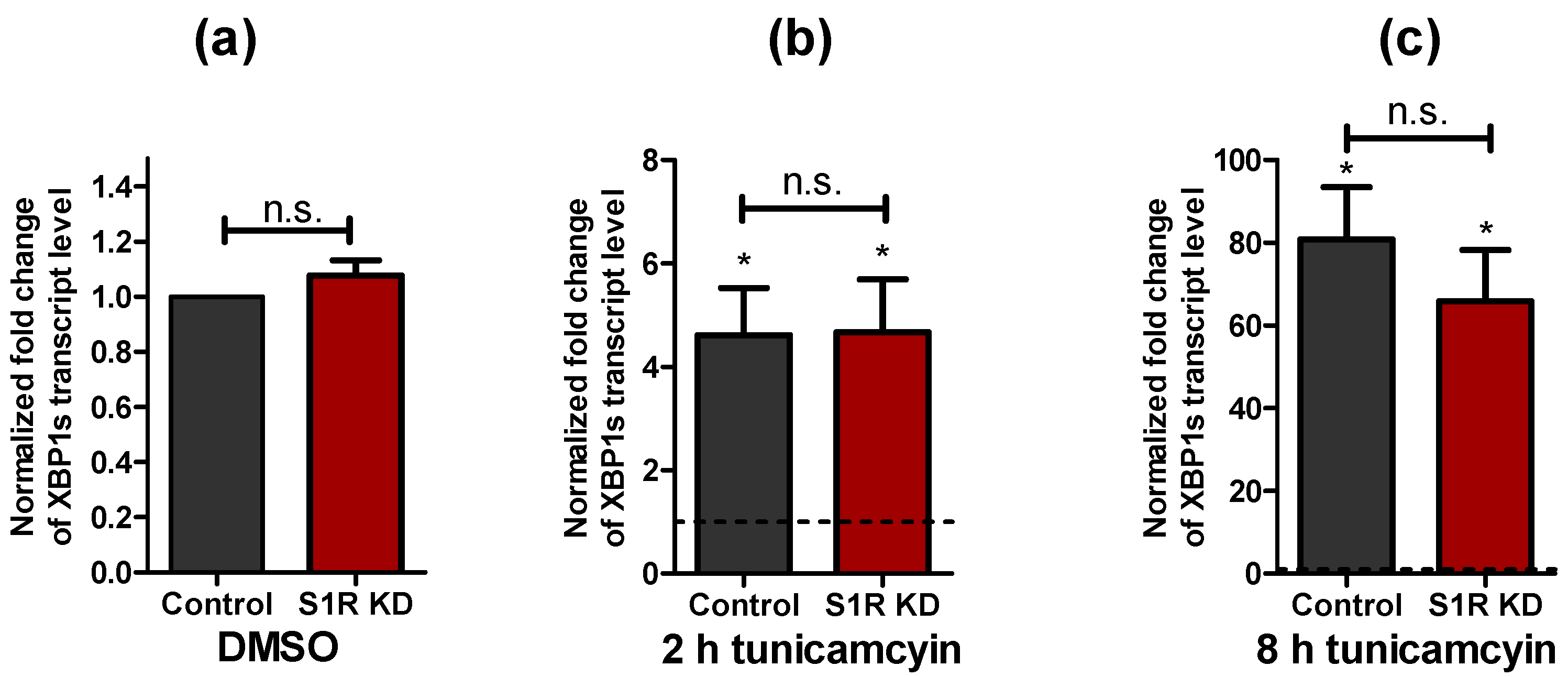
2.1. XBP1 Splicing Increases during Early ER Stress and Is Not Affected by Sigma-1 Receptor Knock-Down
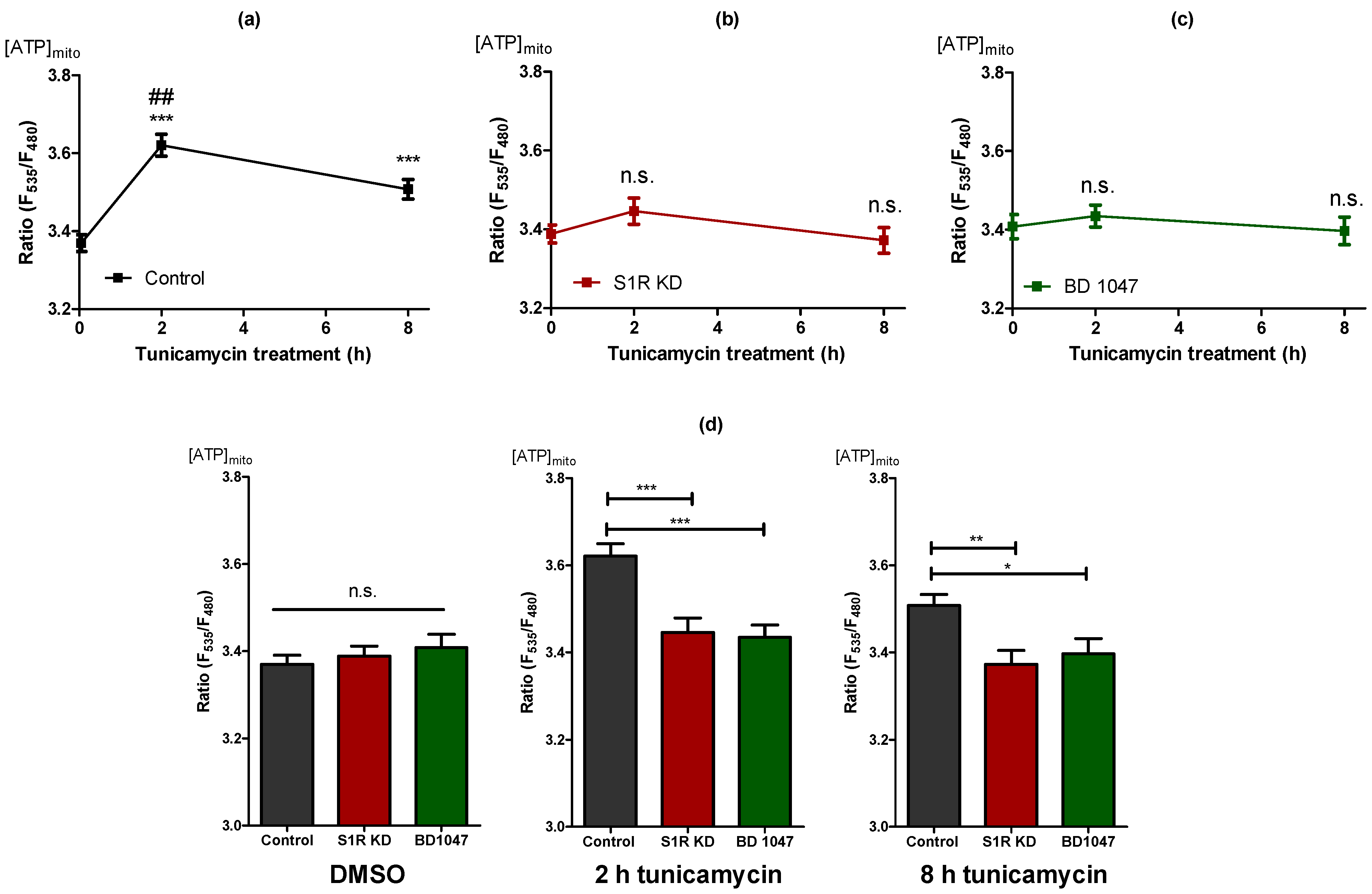
2.2. Sigma-1 Receptor Is Essential for Increased Mitochondrial ATP Level during Early ER Stress
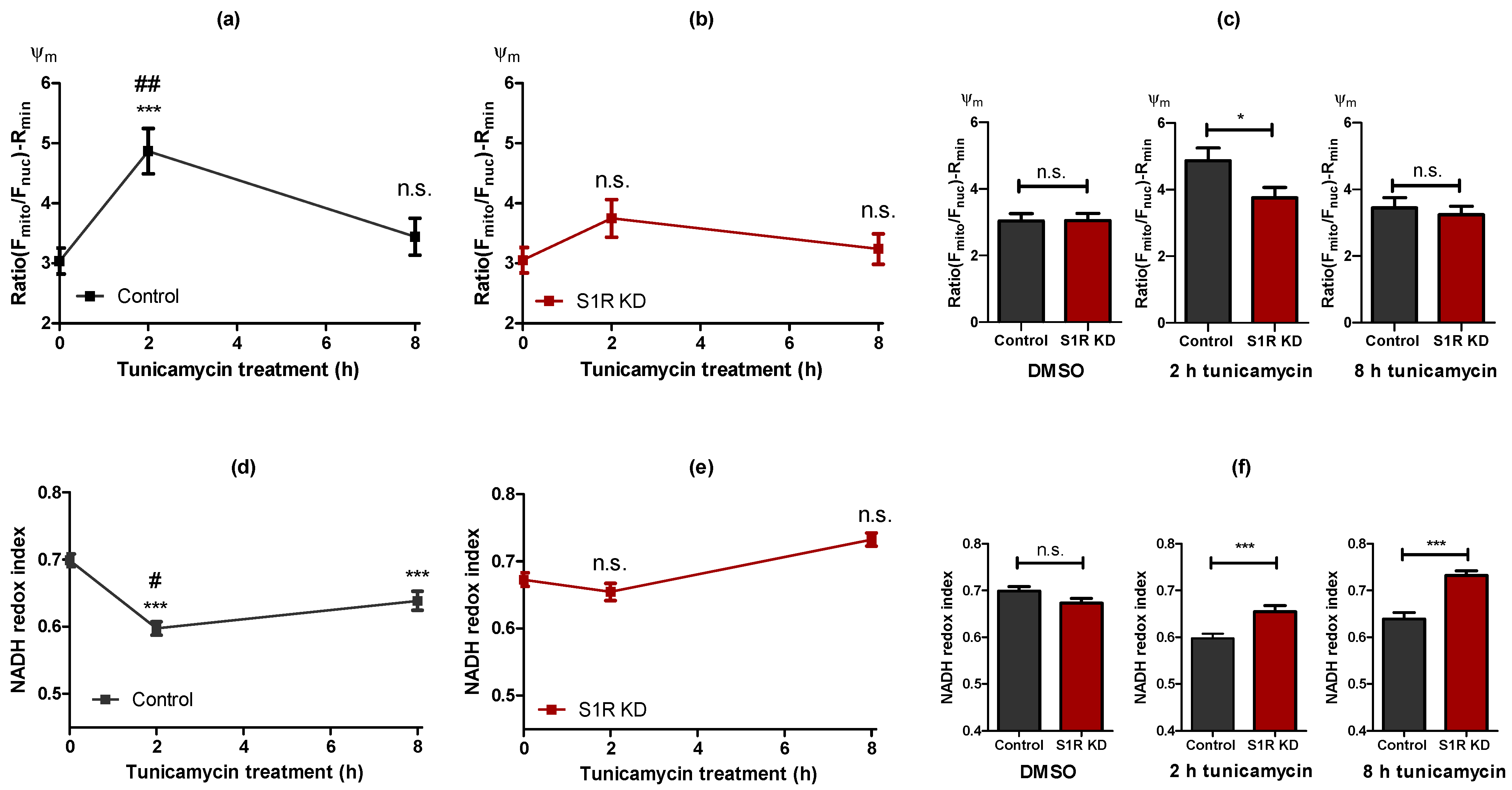
2.3. Sigma-1 Receptor Is Promoting Mitochondrial Hyperpolarisation and NADH Consumption during Early ER Stress
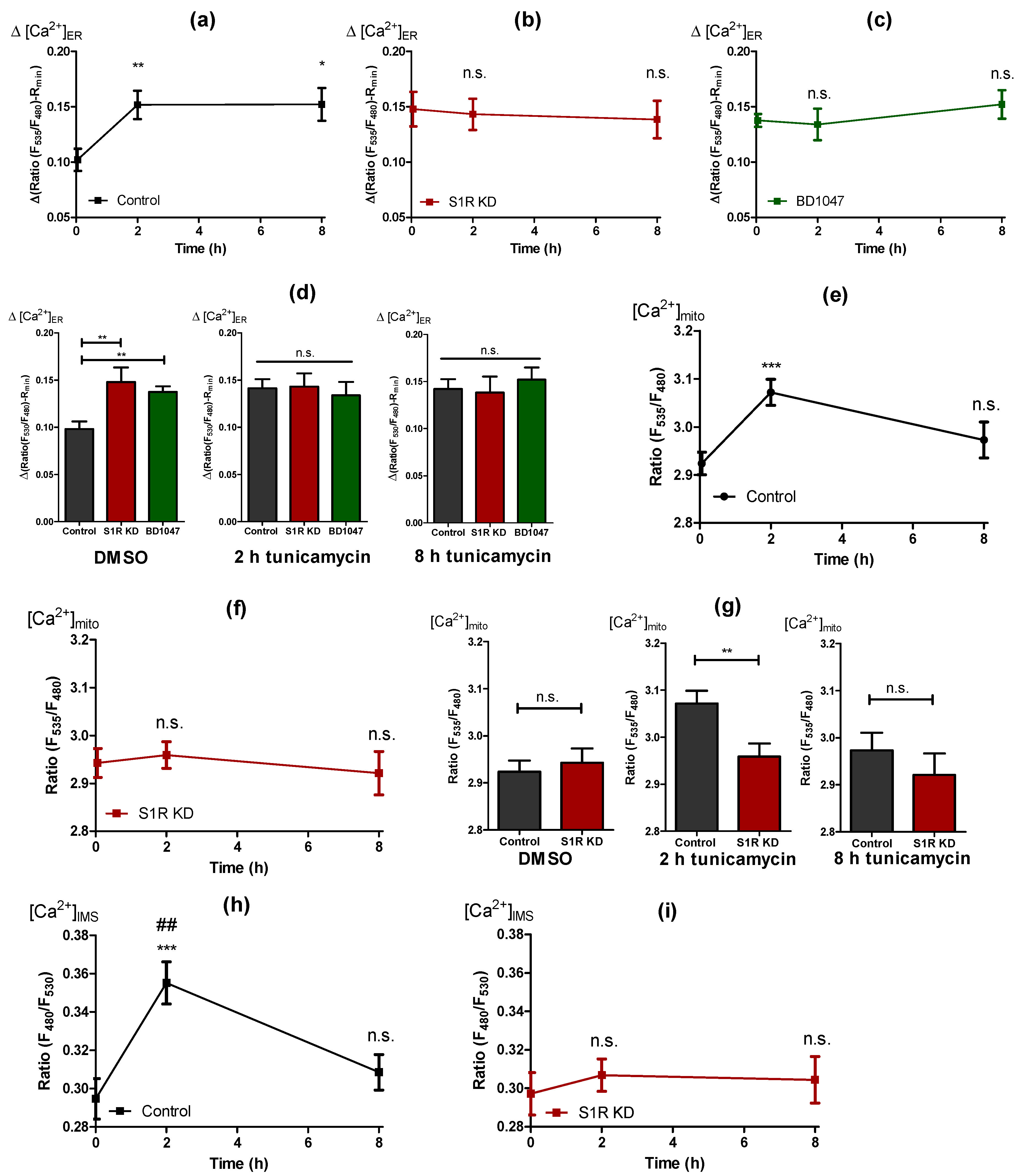
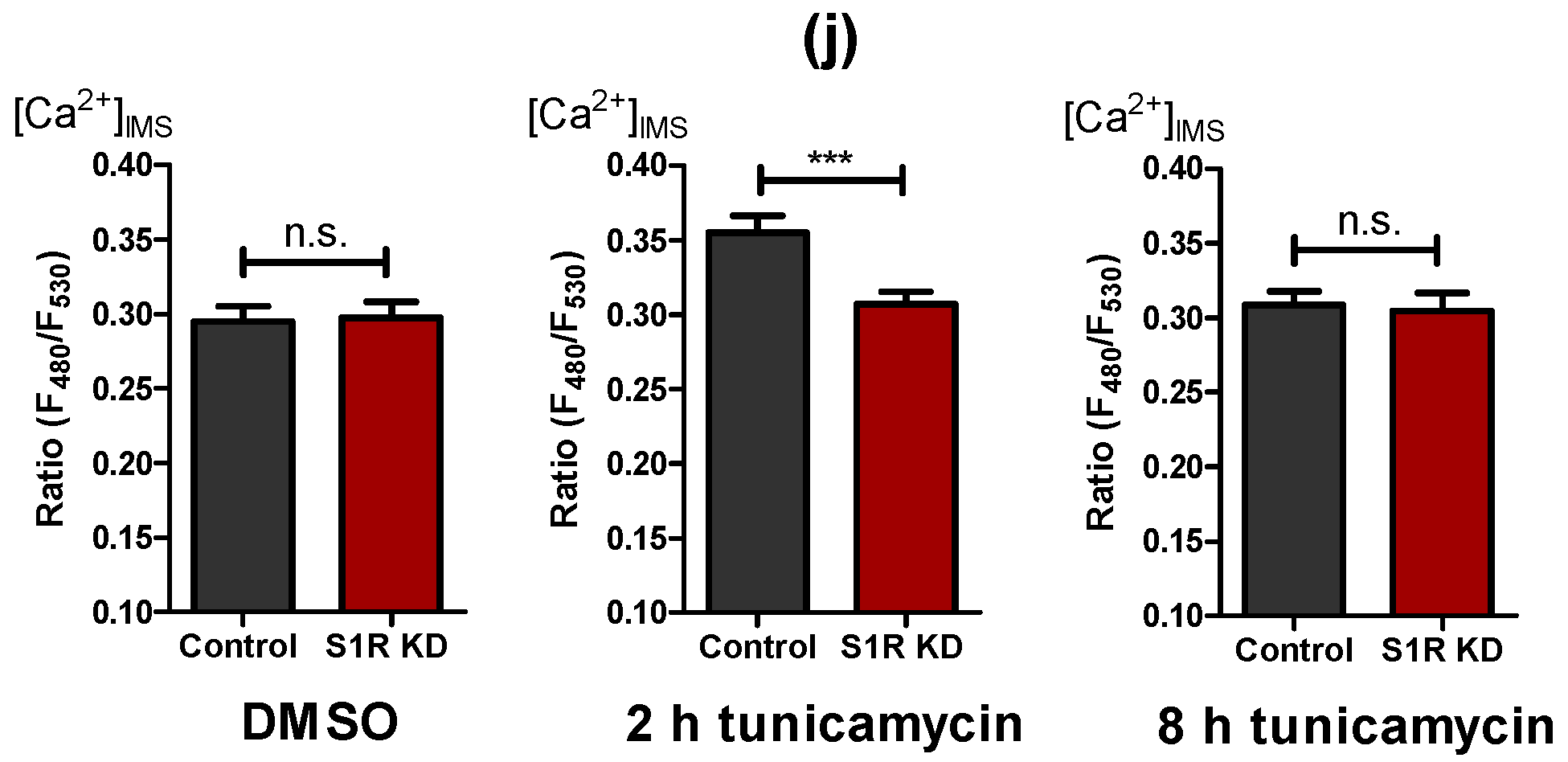
2.4. Early ER Stress Gives Rise to an Increased ER Ca2+ Flux That Is Directed towards Mitochondria by Sigma-1 Receptor
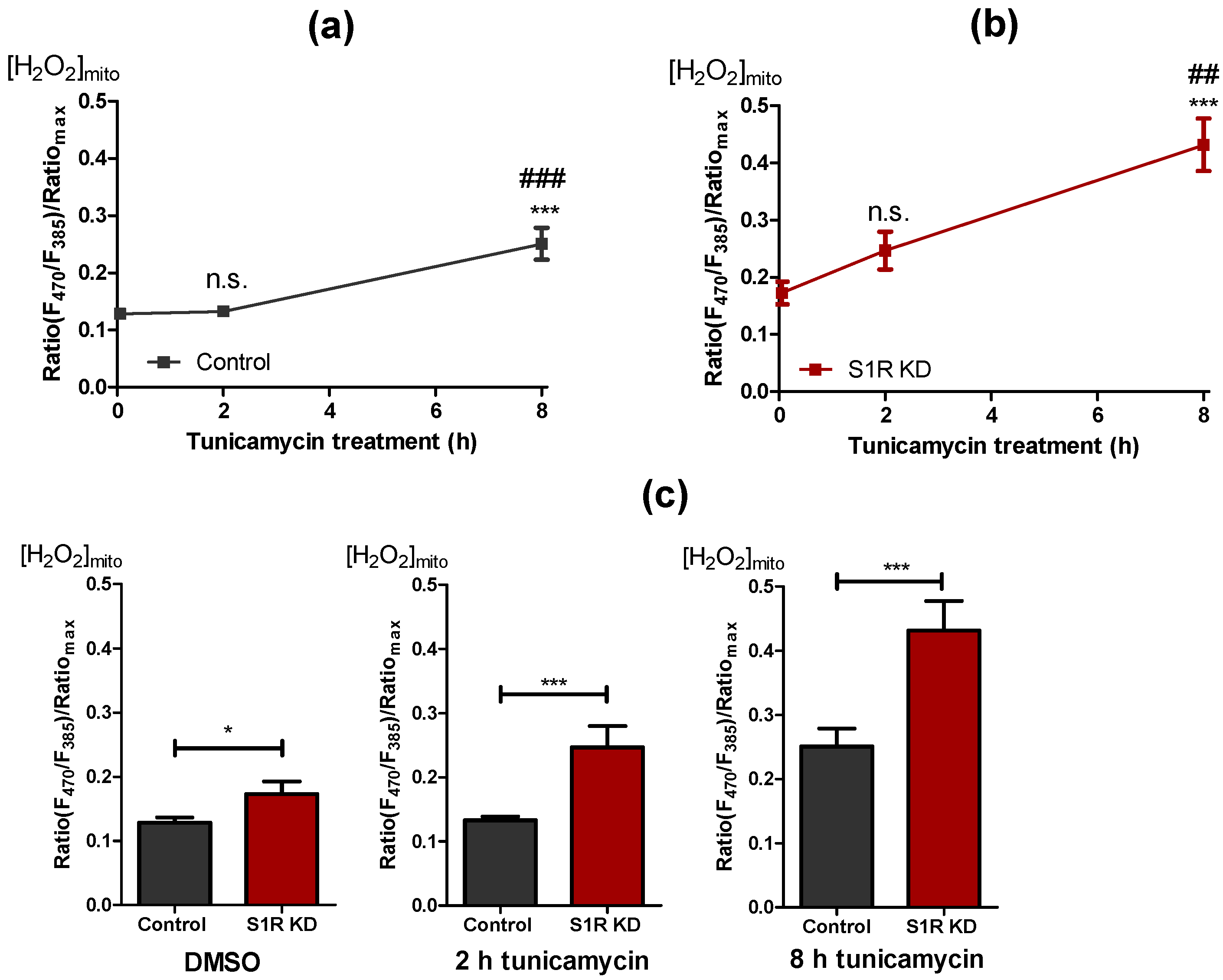
2.5. Sigma-1 Receptor Protects against Increased Mitochondrial ROS Production during ER Stress
3. Discussion
4. Materials and Methods
| Abbreviation | Expanded Version | |
| ER | Endoplasmic reticulum | |
| UPR | Unfolded protein response | |
| S1R | Sigma-1 Receptor | |
| PERK | Double-stranded RNA-dependent protein kinase (PRK)-like ER kinase | |
| ATF6 | Activating transcription factor 6 | |
| IRE1 | Inositol requiring enzyme 1 | |
| ERAD | Endoplasmic-reticulum-associated protein degradation | |
| MAMs | Mitochondria-associated ER membranes | |
| IP3R3 | Inositol 1,4,5-trisphosphate receptor | |
| XBP1 | X-box binding protein 1 | |
| KD | Knock-down | |
| siRNA | Small interfering RNA | |
| GFP | Green fluorescent protein | |
| DMSO | Dimethyl sulfoxide | |
| ATP | Adenosine triphosphate | |
| FRET | Förster resonance energy transfer | |
| Ψm | Mitochondrial membrane potential | |
| NADH | Nicotinamide adenine dinucleotide | |
| IMS | Mitochondrial inter-membrane space | |
| ROS | Reactive oxygen species | |
| SERCA | Sarco(endo)plasmic reticulum Ca2+-ATPase | |
| Sensor | Definition | Reference |
| mtAT1.03 | Genetically encoded mitochondrial matrix targeted FRET-based ATP biosensor | [14] |
| TMRM | Tetramethylrhodamine methyl ester | |
| D1ER | Genetically encoded ER targeted FRET-based Ca2+ biosensor | [17] |
| 4mtD3cpv | Genetically encoded mitochondrial matrix targeted FRET-based Ca2+ biosensor | [18] |
| IMS-GEM-GECO | Genetically encoded mitochondrial IMS targeted ratiometric Ca2+ biosensor | [19,20] |
| mitoHyper7 | Genetically encoded mitochondrial matrix targeted ratiometric ROS biosensor | [24] |
4.1. Cell Culture and Transfection
4.2. Quantitative PCR and XBP1 Splicing
4.3. Live Cell Imaging
4.4. Mitochondrial ATP, Membrane Potential and NADH Redox Index Measurements
4.5. ER Ca2+ Measurements
4.6. ER-Mitochondria Co-Localization
4.7. Mitochondrial and IMS Ca2+ Measurements
4.8. Mitochondrial ROS Measurements
4.9. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Yong, J.; Bischof, H.; Burgstaller, S.; Siirin, M.; Murphy, A.; Malli, R.; Kaufman, R.J. Mitochondria supply ATP to the ER through a mechanism antagonized by cytosolic Ca2+. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Muñoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Deniaud, A.; El Dein, O.S.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; A Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2007, 27, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.-P. Sigma-1 Receptor Chaperone at the ER-Mitochondrion Interface Mediates the Mitochondrion-ER-Nucleus Signaling for Cellular Survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nat. Cell Biol. 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Yoon, S.-B.; Park, Y.-H.; Choi, S.-A.; Yang, H.-J.; Jeong, P.-S.; Cha, J.-J.; Lee, S.; Lee, S.H.; Lee, J.-H.; Sim, B.-W.; et al. Real-time PCR quantification of spliced X-box binding protein 1 (XBP1) using a universal primer method. PLoS ONE 2019, 14, e0219978. [Google Scholar] [CrossRef] [Green Version]
- Imamura, H.; Nhat, K.P.H.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. USA 2009, 106, 15651–15656. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, R.R.; Bowen, W.D.; Tom, M.A.; Vo, V.N.; Truong, D.D.; De Costa, B.R. Characterization of two novel σ receptor ligands: Antidystonic effects in rats suggest σ receptor antagonism. Eur. J. Pharmacol. 1995, 280, 301–310. [Google Scholar] [CrossRef]
- Bartolomé, F.; Abramov, A.Y. Measurement of Mitochondrial NADH and FAD Autofluorescence in Live Cells. Methods Mol. Biol. 2015, 1264, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.E.; Jin, C.; Reed, J.C.; Tsien, R.Y. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. USA 2004, 101, 17404–17409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, A.E.; Giacomello, M.; Kortemme, T.; Hires, S.A.; Lev-Ram, V.; Baker, D.; Tsien, R.Y. Ca2+ Indicators Based on Computationally Redesigned Calmodulin-Peptide Pairs. Chem. Biol. 2006, 13, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldeck-Weiermair, M.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Ramadani-Muja, J.; Ziomek, G.; Klec, C.; Burgstaller, S.; Bischof, H.; Depaoli, M.R.; Eroglu, E.; et al. Development and Application of Sub-Mitochondrial Targeted Ca2+ Biosensors. Front. Cell. Neurosci. 2019, 13, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.-F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An expanded palette of genetically encoded Ca2+ indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandary, B.; Marahatta, A.; Kim, H.-R.; Chae, H.-J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef]
- Bjorkman, S.H.; Pereira, R.O. The Interplay Between Mitochondrial Reactive Oxygen Species, Endoplasmic Reticulum Stress, and Nrf2 Signaling in Cardiometabolic Health. Antioxid. Redox Signal. 2021, 2020, 8220. [Google Scholar] [CrossRef]
- Pal, A.; Fontanilla, D.; Gopalakrishnan, A.; Chae, Y.-K.; Markley, J.L.; Ruoho, A.E. The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur. J. Pharmacol. 2012, 682, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Pak, V.V.; Ezerina, D.; Lyublinskaya, O.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Gache, S.A.M.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab. 2020, 31, 642–653.e6. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Gottschalk, B.; Sokolowski, A.A.; Malli, R.; Graier, W.F. Dynamic Control of Mitochondrial Ca2+ Levels as a Survival Strategy of Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Van Coppenolle, F.; Lehen’Kyi, V.; Beck, B.; Skryma, R.; Prevarskaya, N. Passive calcium leak via translocon is a first step for iPLA 2 -pathway regulated store operated channels activation. FASEB J. 2006, 20, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Chami, M.; Oules, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Brechot, P.P. Role of SERCA1 Truncated Isoform in the Proapoptotic Calcium Transfer from ER to Mitochondria during ER Stress. Mol. Cell 2008, 32, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Srivats, S.; Balasuriya, D.; Pasche, M.; Vistal, G.; Edwardson, J.M.; Taylor, C.; Murrell-Lagnado, R.D. Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J. Cell Biol. 2016, 213, 65–79. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koshenov, Z.; Oflaz, F.E.; Hirtl, M.; Pilic, J.; Bachkoenig, O.A.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Rost, R.; Malli, R.; Graier, W.F. Sigma-1 Receptor Promotes Mitochondrial Bioenergetics by Orchestrating ER Ca2+ Leak during Early ER Stress. Metabolites 2021, 11, 422. https://doi.org/10.3390/metabo11070422
Koshenov Z, Oflaz FE, Hirtl M, Pilic J, Bachkoenig OA, Gottschalk B, Madreiter-Sokolowski CT, Rost R, Malli R, Graier WF. Sigma-1 Receptor Promotes Mitochondrial Bioenergetics by Orchestrating ER Ca2+ Leak during Early ER Stress. Metabolites. 2021; 11(7):422. https://doi.org/10.3390/metabo11070422
Chicago/Turabian StyleKoshenov, Zhanat, Furkan E. Oflaz, Martin Hirtl, Johannes Pilic, Olaf A. Bachkoenig, Benjamin Gottschalk, Corina T. Madreiter-Sokolowski, Rene Rost, Roland Malli, and Wolfgang F. Graier. 2021. "Sigma-1 Receptor Promotes Mitochondrial Bioenergetics by Orchestrating ER Ca2+ Leak during Early ER Stress" Metabolites 11, no. 7: 422. https://doi.org/10.3390/metabo11070422