
Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era


Abstract
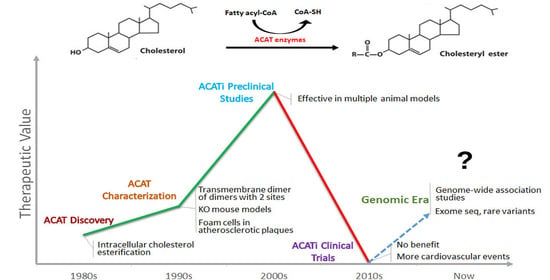
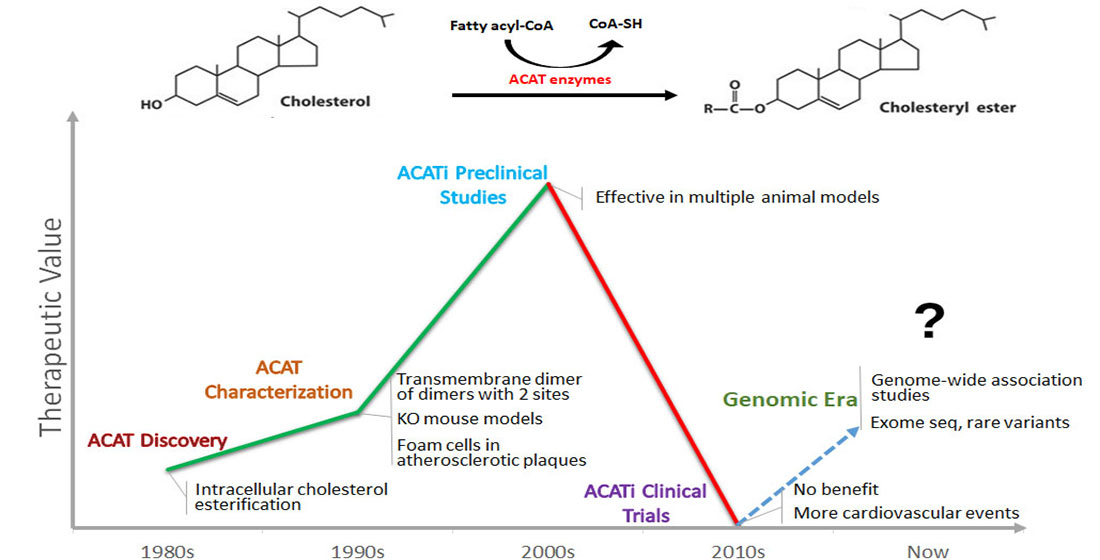
1. Introduction
2. The Structure of ACAT1
3. The Role of ACAT in Lipid Metabolism and Atherosclerosis Derived from Knockout Mouse Studies
4. ACAT Inhibitors in Pre-Clinical Studies
- Rabbit models
- Mouse models
- Adverse effects of ACAT inhibitors
5. Clinical Trials of ACAT Inhibitors
6. ACAT Genetics and Genomics
6.1. Mouse Soat1 Genetics
6.2. Human SOAT1/2 Genes and Expression
6.3. Human SOAT1/2 Common and Rare Genetic Variants and Disease Association
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chang, T.Y.; Li, B.L.; Chang, C.C.; Urano, Y. Acyl-coenzyme A:cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Heider, J.G.; Chang, T.Y. Isolation and characterization of Chinese hamster ovary cell mutants deficient in acyl-coenzyme A:cholesterol acyltransferase activity. J. Biol. Chem. 1988, 263, 274–282. [Google Scholar] [CrossRef]
- Pavanello, C.; Calabresi, L. Genetic, biochemical, and clinical features of LCAT deficiency: Update for 2020. Curr. Opin. Lipidol. 2020, 31, 232–237. [Google Scholar] [CrossRef]
- Anderson, R.A.; Joyce, C.; Davis, M.; Reagan, J.W.; Clark, M.; Shelness, G.S.; Rudel, L.L. Identification of a form of acyl-CoA:cholesterol acyltransferase specific to liver and intestine in nonhuman primates. J. Biol. Chem. 1998, 273, 26747–26754. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Chang, C.C.; Lin, S.; Yu, C.; Li, B.L.; Miyazaki, A. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and -2. Curr. Opin. Lipidol. 2001, 12, 289–296. [Google Scholar] [CrossRef]
- Cases, S.; Novak, S.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Welch, C.B.; Lusis, A.J.; Spencer, T.A.; Krause, B.R.; et al. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J. Biol. Chem. 1998, 273, 26755–26764. [Google Scholar] [CrossRef]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K. A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochem. Sci. 2000, 25, 111–112. [Google Scholar] [CrossRef]
- Rogers, M.A.; Liu, J.; Song, B.L.; Li, B.L.; Chang, C.C.; Chang, T.Y. Acyl-CoA:cholesterol acyltransferases (ACATs/SOATs): Enzymes with multiple sterols as substrates and as activators. J. Steroid Biochem. Mol. Biol. 2015, 151, 102–107. [Google Scholar] [CrossRef]
- Chang, C.; Dong, R.; Miyazaki, A.; Sakashita, N.; Zhang, Y.; Liu, J.; Guo, M.; Li, B.L.; Chang, T.Y. Human acyl-CoA:cholesterol acyltransferase (ACAT) and its potential as a target for pharmaceutical intervention against atherosclerosis. Acta Biochim. Biophys. Sin. 2006, 38, 151–156. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Chang, C.C.; Chang, T.Y. Isolation of Chinese hamster ovary cell lines expressing human acyl-coenzyme A/cholesterol acyltransferase activity. J. Cell Biol. 1989, 108, 2201–2210. [Google Scholar] [CrossRef]
- Chang, C.C.; Huh, H.Y.; Cadigan, K.M.; Chang, T.Y. Molecular cloning and functional expression of human acyl-coenzyme A:cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J. Biol. Chem. 1993, 268, 20747–20755. [Google Scholar] [CrossRef]
- Chang, C.C.; Noll, W.W.; Nutile-McMenemy, N.; Lindsay, E.A.; Baldini, A.; Chang, W.; Chang, T.Y. Localization of acyl coenzyme A:cholesterol acyltransferase gene to human chromosome 1q25. Somat. Cell Mol. Genet. 1994, 20, 71–74. [Google Scholar] [CrossRef]
- Chang, C.C.; Lee, C.Y.; Chang, E.T.; Cruz, J.C.; Levesque, M.C.; Chang, T.Y. Recombinant acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) purified to essential homogeneity utilizes cholesterol in mixed micelles or in vesicles in a highly cooperative manner. J. Biol. Chem. 1998, 273, 35132–35141. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, C.; Liu, J.; Spencer, T.A.; Chang, C.C.Y.; Chang, T.-Y. Cholesterol is superior to 7-ketocholesterol or 7 alpha-hydroxycholesterol as an allosteric activator for acyl-coenzyme A:cholesterol acyltransferase 1. J. Biol. Chem. 2003, 278, 11642–11647. [Google Scholar] [CrossRef]
- Rogers, M.A.; Liu, J.; Kushnir, M.M.; Bryleva, E.; Rockwood, A.L.; Meikle, A.W.; Shapiro, D.; Vaisman, B.L.; Remaley, A.T.; Chang, C.C.Y.; et al. Cellular pregnenolone esterification by acyl-CoA:cholesterol acyltransferase. J. Biol. Chem. 2012, 287, 17483–17492. [Google Scholar] [CrossRef]
- Chang, C.C.; Miyazaki, A.; Dong, R.; Kheirollah, A.; Yu, C.; Geng, Y.; Higgs, H.N.; Chang, T.Y. Purification of recombinant acyl-coenzyme A:cholesterol acyltransferase 1 (ACAT1) from H293 cells and binding studies between the enzyme and substrates using difference intrinsic fluorescence spectroscopy. Biochemistry 2010, 49, 9957–9963. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seo, T.; Oelkers, P.M.; Giattina, M.R.; Worgall, T.S.; Sturley, S.L.; Deckelbaum, R.J. Differential modulation of ACAT1 and ACAT2 transcription and activity by long chain free fatty acids in cultured cells. Biochemistry 2001, 40, 4756–4762. [Google Scholar] [CrossRef] [PubMed]
- Temel, R.E.; Gebre, A.K.; Parks, J.S.; Rudel, L.L. Compared with Acyl-CoA:cholesterol O-acyltransferase (ACAT) 1 and lecithin:cholesterol acyltransferase, ACAT2 displays the greatest capacity to differentiate cholesterol from sitosterol. J. Biol. Chem. 2003, 278, 47594–47601. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Zhao, X.; Yan, R.; Yao, X.; Gao, S.; Sun, X.; Du, X.; Yang, H.; Wong, C.C.L.; Yan, N. Structural basis for catalysis and substrate specificity of human ACAT1. Nature 2020, 581, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Meiner, V.L.; Cases, S.; Myers, H.M.; Sande, E.R.; Bellosta, S.; Schambelan, M.; Pitas, R.E.; McGuire, J.; Herz, J.; Farese, R.V.J. Disruption of the acyl-CoA:cholesterol acyltransferase gene in mice: Evidence suggesting multiple cholesterol esterification enzymes in mammals. Proc. Natl. Acad. Sci. USA 1996, 93, 14041–14046. [Google Scholar] [CrossRef]
- Huang, L.H.; Gui, J.; Artinger, E.; Craig, R.; Berwin, B.L.; Ernst, P.A.; Chang, C.C.; Chang, T.Y. Acat1 gene ablation in mice increases hematopoietic progenitor cell proliferation in bone marrow and causes leukocytosis. Arter. Thromb. Vasc. Biol. 2013, 33, 2081–2087. [Google Scholar] [CrossRef] [PubMed]
- Buhman, K.K.; Accad, M.; Novak, S.; Choi, R.S.; Wong, J.S.; Hamilton, R.L.; Turley, S.; Farese, R.V.J. Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat. Med. 2000, 6, 1341–1347. [Google Scholar] [CrossRef]
- Repa, J.J.; Buhman, K.K.; Farese, R.V.J.; Dietschy, J.M.; Turley, S.D. ACAT2 deficiency limits cholesterol absorption in the cholesterol-fed mouse: Impact on hepatic cholesterol homeostasis. Hepatology 2004, 40, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Accad, M.; Smith, S.J.; Newland, D.L.; Sanan, D.A.; King, L.E.J.; Linton, M.F.; Fazio, S.; Farese, R.V.J. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J. Clin. Investig. 2000, 105, 711–719. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fazio, S.; Major, A.S.; Swift, L.L.; Gleaves, L.A.; Accad, M.; Linton, M.F.; Farese, R.V.J. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J. Clin. Investig. 2001, 107, 163–171. [Google Scholar] [CrossRef]
- Yagyu, H.; Kitamine, T.; Osuga, J.I.; Tozawa, R.I.; Chen, Z.; Kaji, Y.; Oka, T.; Perrey, S.; Tamura, Y.; Ohashi, K.; et al. Absence of ACAT-1 attenuates atherosclerosis but ’causes dry eye and cutaneous xanthomatosis mice with congenital hyperlipidemia. J. Biol. Chem. 2000, 275, 21324–21330. [Google Scholar] [CrossRef]
- Huang, L.H.; Melton, E.M.; Li, H.; Sohn, P.; Rogers, M.A.; Mulligan-Kehoe, M.J.; Fiering, S.N.; Hickey, W.F.; Chang, C.C.; Chang, T.Y. Myeloid Acyl-CoA:Cholesterol Acyltransferase 1 Deficiency Reduces Lesion Macrophage Content and Suppresses Atherosclerosis Progression. J. Biol. Chem. 2016, 291, 6232–6244. [Google Scholar] [CrossRef] [PubMed]
- Melton, E.M.; Li, H.; Benson, J.; Sohn, P.; Huang, L.-H.; Song, B.-L.; Li, B.-L.; Chang, C.C.Y.; Chang, T.-Y. Myeloid Acat1/Soat1 KO attenuates pro-inflammatory responses in macrophages and protects against atherosclerosis in a model of advanced lesions. J. Biol. Chem. 2019, 294, 15836–15849. [Google Scholar] [CrossRef]
- Willner, E.L.; Tow, B.; Buhman, K.K.; Wilson, M.; Sanan, D.A.; Rudel, L.L.; Farese, R.V.J. Deficiency of acyl CoA:cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1262–1267. [Google Scholar] [CrossRef]
- Kusunoki, J.; Aragane, K.; Kitamine, T.; Higashinakagawa, S.; Kase, N.; Yamaura, T.; Ohnishi, H. Hypocholesterolemic action and prevention of cholesterol absorption via the gut by F-1394, a potent acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor, in cholesterol diet-fed rats. Jpn. J. Pharmacol. 1995, 69, 53–60. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chiwata, T.; Aragane, K.; Fujinami, K.; Kojima, K.; Ishibashi, S.; Yamada, N.; Kusunoki, J. Direct effect of an acyl-CoA:cholesterol acyltransferase inhibitor, F-1394, on atherosclerosis in apolipoprotein E and low density lipoprotein receptor double knockout mice. Br. J. Pharmacol. 2001, 133, 1005–1012. [Google Scholar] [CrossRef]
- Aragane, K.; Fujinami, K.; Kojima, K.; Kusunoki, J. ACAT inhibitor F-1394 prevents intimal hyperplasia induced by balloon injury in rabbits. J. Lipid Res. 2001, 42, 480–488. [Google Scholar] [CrossRef]
- Kusunoki, J.; Hansoty, D.K.; Aragane, K.; Fallon, J.T.; Badimon, J.J.; Fisher, E.A. Acyl-CoA:cholesterol acyltransferase inhibition reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 2001, 103, 2604–2609. [Google Scholar] [CrossRef]
- Rong, J.X.; Blachford, C.; Feig, J.E.; Bander, I.; Mayne, J.; Kusunoki, J.; Miller, C.; Davis, M.; Wilson, M.; Dehn, S.; et al. ACAT inhibition reduces the progression of preexisting, advanced atherosclerotic mouse lesions without plaque or systemic toxicity. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.; Ogando, Y.; Nikain, C.; Quezada, A.; Qian, K.; Vaisar, T.; Fisher, E.A. Short-Term Acyl-CoA:Cholesterol Acyltransferase Inhibition, Combined with Apoprotein A1 Overexpression, Promotes Atherosclerosis Inflammation Resolution in Mice. Mol. Pharmacol. 2021, 99, 175–183. [Google Scholar] [CrossRef]
- Ramharack, R.; Spahr, M.A.; Sekerke, C.S.; Stanfield, R.L.; Bousley, R.F.; Lee, H.T.; Krause, B.K. CI-1011 lowers lipoprotein(a) and plasma cholesterol concentrations in chow-fed cynomolgus monkeys. Atherosclerosis 1998, 136, 79–87. [Google Scholar] [CrossRef]
- Burnett, J.R.; Wilcox, L.J.; Telford, D.E.; Kleinstiver, S.J.; Barrett, P.H.; Newton, R.S.; Huff, M.W. Inhibition of ACAT by avasimibe decreases both VLDL and LDL apolipoprotein B production in miniature pigs. J. Lipid Res. 1999, 40, 1317–1327. [Google Scholar] [CrossRef]
- Bocan, T.M.; Krause, B.R.; Rosebury, W.S.; Mueller, S.B.; Lu, X.; Dagle, C.; Major, T.; Lathia, C.; Lee, H. The ACAT inhibitor avasimibe reduces macrophages and matrix metalloproteinase expression in atherosclerotic lesions of hypercholesterolemic rabbits. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 70–79. [Google Scholar] [CrossRef][Green Version]
- Robertson, D.G.; Breider, M.A.; Milad, M.A. Preclinical safety evaluation of avasimibe in beagle dogs: An ACAT inhibitor with minimal adrenal effects. Toxicol. Sci. 2001, 59, 324–334. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Delsing, D.J.; Offerman, E.H.; van Duyvenvoorde, W.; van Der Boom, H.; de Wit, E.C.; Gijbels, M.J.; van Der Laarse, A.; Jukema, J.W.; Havekes, L.M.; Princen, H.M. Acyl-CoA:cholesterol acyltransferase inhibitor avasimibe reduces atherosclerosis in addition to its cholesterol-lowering effect in ApoE*3-Leiden mice. Circulation 2001, 103, 1778–1786. [Google Scholar] [CrossRef]
- Burnett, J.R.; Telford, D.E.; Barrett, P.H.R.; Huff, M.W. The ACAT inhibitor avasimibe increases the fractional clearance rate of postprandial triglyceride-rich lipoproteins in miniature pigs. Biochim. Biophys. Acta 2005, 1738, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Miyazaki, A.; Kasanuki, N.; Ito, K.; Ubukata, N.; Koieyama, T.; Kitayama, K.; Tanimoto, T.; Maeda, N.; Inaba, T. ACAT inhibitor pactimibe sulfate (CS-505) reduces and stabilizes atherosclerotic lesions by cholesterol-lowering and direct effects in apolipoprotein E-deficient mice. Atherosclerosis 2007, 190, 239–247. [Google Scholar] [CrossRef]
- Worthley, S.G.; Helft, G.; Corti, R.; Worthley, M.I.; Chew, D.P.; Fayad, Z.A.; Zaman, A.G.; Fallon, J.T.; Fuster, V.; Badimon, J.J. Statin therapy alone and in combination with an acyl-CoA:cholesterol O-acyltransferase inhibitor on experimental atherosclerosis. Pathophysiol. Haemost. Thromb. 2007, 36, 9–17. [Google Scholar] [CrossRef]
- Kitayama, K.; Koga, T.; Maeda, N.; Inaba, T.; Fujioka, T. Pactimibe stabilizes atherosclerotic plaque through macrophage acyl-CoA:cholesterol acyltransferase inhibition in WHHL rabbits. Eur. J. Pharmacol. 2006, 539, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, K.; Tanimoto, T.; Koga, T.; Terasaka, N.; Fujioka, T.; Inaba, T. Importance of acyl-coenzyme A:cholesterol acyltransferase 1/2 dual inhibition for anti-atherosclerotic potency of pactimibe. Eur. J. Pharmacol. 2006, 540, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, K.; Koga, T.; Inaba, T.; Fujioka, T. Multiple mechanisms of hypocholesterolemic action of pactimibe, a novel acyl-coenzyme A:cholesterol acyltransferase inhibitor. Eur. J. Pharmacol. 2006, 543, 123–132. [Google Scholar] [CrossRef]
- Yoshinaka, Y.; Shibata, H.; Kobayashi, H.; Kuriyama, H.; Shibuya, K.; Tanabe, S.; Watanabe, T.; Miyazaki, A. A selective ACAT-1 inhibitor, K-604, stimulates collagen production in cultured smooth muscle cells and alters plaque phenotype in apolipoprotein E-knockout mice. Atherosclerosis 2010, 213, 85–91. [Google Scholar] [CrossRef]
- Bocan, T.M.; Mueller, S.B.; Uhlendorf, P.D.; Newton, R.S.; Krause, B.R. Comparison of CI-976, an ACAT inhibitor, and selected lipid-lowering agents for antiatherosclerotic activity in iliac-femoral and thoracic aortic lesions. A biochemical, morphological, and morphometric evaluation. Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 1830–1843. [Google Scholar] [CrossRef]
- Krause, B.R.; Bousley, R.F.; Kieft, K.A.; Stanfield, R.L. Effect of the ACAT inhibitor CI-976 on plasma cholesterol concentrations and distribution in hamsters fed zero- and low-cholesterol diets. Clin. Biochem. 1992, 25, 371–377. [Google Scholar] [CrossRef]
- Krause, B.R.; Anderson, M.; Bisgaier, C.L.; Bocan, T.; Bousley, R.; DeHart, P.; Essenburg, A.; Hamelehle, K.; Homan, R.; Kieft, K. In vivo evidence that the lipid-regulating activity of the ACAT inhibitor CI-976 in rats is due to inhibition of both intestinal and liver ACAT. J. Lipid Res. 1993, 34, 279–294. [Google Scholar] [CrossRef]
- Bocan, T.M.; Mueller, S.B.; Uhlendorf, P.D.; Brown, E.Q.; Mazur, M.J.; Black, A.E. Inhibition of acyl-CoA cholesterol O-acyltransferase reduces the cholesteryl ester enrichment of atherosclerotic lesions in the Yucatan micropig. Atherosclerosis 1993, 99, 175–186. [Google Scholar] [CrossRef]
- Krause, B.R.; Pape, M.E.; Kieft, K.; Auerbach, B.; Bisgaier, C.L.; Homan, R.; Newton, R.S. ACAT inhibition decreases LDL cholesterol in rabbits fed a cholesterol-free diet. Marked changes in LDL cholesterol without changes in LDL receptor mRNA abundance. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 598–604. [Google Scholar] [CrossRef]
- Bocan, T.M.; Mueller, S.B.; Brown, E.Q.; Lee, P.; Bocan, M.J.; Rea, T.; Pape, M.E. HMG-CoA reductase and ACAT inhibitors act synergistically to lower plasma cholesterol and limit atherosclerotic lesion development in the cholesterol-fed rabbit. Atherosclerosis 1998, 139, 21–30. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Liang, K.H. Acyl-coenzyme A:cholesterol acyltransferase inhibition ameliorates proteinuria, hyperlipidemia, lecithin-cholesterol acyltransferase, SRB-1, and low-denisty lipoprotein receptor deficiencies in nephrotic syndrome. Circulation 2004, 110, 419–425. [Google Scholar] [CrossRef]
- Ikenoya, M.; Yoshinaka, Y.; Kobayashi, H.; Kawamine, K.; Shibuya, K.; Sato, F.; Sawanobori, K.; Watanabe, T.; Miyazaki, A. A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 2007, 191, 290–297. [Google Scholar] [CrossRef]
- Krause, B.R.; Black, A.; Bousley, R.; Essenburg, A.; Cornicelli, J.; Holmes, A.; Homan, R.; Kieft, K.; Sekerke, C.; Shaw-Hes, M.K. Divergent pharmacologic activities of PD 132301-2 and CL 277,082, urea inhibitors of acyl-CoA:cholesterol acyltransferase. J. Pharmacol. Exp. Ther. 1993, 267, 734–743. [Google Scholar] [PubMed]
- Azuma, Y.; Date, K.; Ohno, K.; Matsushiro, S.; Nobuhara, Y.; Yamada, T. NTE-122, an acyl-coa:cholesterol acyltransferase inhibitor, prevents the progression of atherogenesis in cholesterol-fed rabbits. Jpn. J. Pharmacol. 2001, 86, 120–123. [Google Scholar] [CrossRef][Green Version]
- Aragane, K.; Kojima, K.; Fujinami, K.; Kamei, J.; Kusunoki, J. Effect of F-1394, an acyl-CoA:cholesterol acyltransferase inhibitor, on atherosclerosis induced by high cholesterol diet in rabbits. Atherosclerosis 2001, 158, 139–145. [Google Scholar] [CrossRef]
- Dominick, M.A.; McGuire, E.J.; Reindel, J.F.; Bobrowski, W.F.; Bocan, T.M.; Gough, A.W. Subacute toxicity of a novel inhibitor of acyl-CoA: Cholesterol acyltransferase in beagle dogs. Fundam. Appl. Toxicol. 1993, 20, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Dominick, M.A.; Bobrowski, W.A.; MacDonald, J.R.; Gough, A.W. Morphogenesis of a zone-specific adrenocortical cytotoxicity in guinea pigs administered PD 132301-2, an inhibitor of acyl-CoA:cholesterol acyltransferase. Toxicol. Pathol. 1993, 21, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Vernetti, L.A.; MacDonald, J.R.; Wolfgang, G.H.; Dominick, M.A.; Pegg, D.G. ATP depletion is associated with cytotoxicity of a novel lipid regulator in guinea pig adrenocortical cells. Toxicol. Appl. Pharmacol. 1993, 118, 30–38. [Google Scholar] [CrossRef]
- Warner, G.J.; Stoudt, G.; Bamberger, M.; Johnson, W.J.; Rothblat, G.H. Cell toxicity induced by inhibition of acyl coenzyme A:cholesterol acyltransferase and accumulation of unesterified cholesterol. J. Biol. Chem. 1995, 270, 5772–5778. [Google Scholar] [CrossRef]
- Kellner-Weibel, G.; Jerome, W.G.; Small, D.M.; Warner, G.J.; Stoltenborg, J.K.; Kearney, M.A.; Corjay, M.H.; Phillips, M.C.; Rothblat, G.H. Effects of intracellular free cholesterol accumulation on macrophage viability: A model for foam cell death. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 423–431. [Google Scholar] [CrossRef]
- Kellner-Weibel, G.; Yancey, P.G.; Jerome, W.G.; Walser, T.; Mason, R.P.; Phillips, M.C.; Rothblat, G.H. Crystallization of free cholesterol in model macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1891–1898. [Google Scholar] [CrossRef]
- Yao, P.M.; Tabas, I. Free cholesterol loading of macrophages induces apoptosis involving the fas pathway. J. Biol. Chem. 2000, 275, 23807–23813. [Google Scholar] [CrossRef]
- Yao, P.M.; Tabas, I. Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J. Biol. Chem. 2001, 276, 42468–42476. [Google Scholar] [CrossRef]
- Devries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.J.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 2005, 171, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Grégoire, J.; L’Allier, P.L.; Anderson, T.J.; Bertrand, O.; Reeves, F.; Title, L.M.; Alfonso, F.; Schampaert, E.; Hassan, A.; et al. Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation 2004, 110, 3372–3377. [Google Scholar] [CrossRef]
- Nissen, S.E.; Tuzcu, E.M.; Brewer, H.B.; Sipahi, I.; Nicholls, S.J.; Ganz, P.; Schoenhagen, P.; Waters, D.D.; Pepine, C.J.; Crowe, T.D.; et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N. Engl. J. Med. 2006, 354, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, M.C.; de Groot, E.; Duivenvoorden, R.; Trip, M.D.; Ose, L.; Maritz, F.J.; Basart, D.C.G.; Kastelein, J.J.P.; Habib, R.; Davidson, M.H.; et al. ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: The CAPTIVATE randomized trial. JAMA 2009, 301, 1131–1139. [Google Scholar] [CrossRef]
- Farese, R.V.J. The nine lives of ACAT inhibitors. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1684–1686. [Google Scholar] [CrossRef][Green Version]
- Sahi, J.; Milad, M.A.; Zheng, X.; Rose, K.A.; Wang, H.; Stilgenbauer, L.; Gilbert, D.; Jolley, S.; Stern, R.H.; LeCluyse, E.L. Avasimibe induces CYP3A4 and multiple drug resistance protein 1 gene expression through activation of the pregnane X receptor. J. Pharmacol. Exp. Ther. 2003, 306, 1027–1034. [Google Scholar] [CrossRef]
- Sahi, J.; Stern, R.H.; Milad, M.A.; Rose, K.A.; Gibson, G.; Zheng, X.; Stilgenbauer, L.; Sadagopan, N.; Jolley, S.; Gilbert, D.; et al. Effects of avasimibe on cytochrome P450 2C9 expression in vitro and in vivo. Drug Metab. Dispos. 2004, 32, 1370–1376. [Google Scholar] [CrossRef]
- ARNESEN, K. Constitutional difference in lipid content of adrenals in two strains of mice and their hybrids. Acta Endocrinol. 1955, 18, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Meiner, V.L.; Welch, C.L.; Cases, S.; Myers, H.M.; Sande, E.; Lusis, A.J.; Farese, R.V.J. Adrenocortical lipid depletion gene (ald) in AKR mice is associated with an acyl-CoA:cholesterol acyltransferase (ACAT) mutation. J. Biol. Chem. 1998, 273, 1064–1069. [Google Scholar] [CrossRef] [PubMed]
- Hai, Q.; Ritchey, B.; Robinet, P.; Alzayed, A.M.; Brubaker, G.; Zhang, J.; Smith, J.D. Quantitative Trait Locus Mapping of Macrophage Cholesterol Metabolism and CRISPR/Cas9 Editing Implicate an ACAT1 Truncation as a Causal Modifier Variant. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Opoku, E.; Traughber, C.A.; Hai, Q.; Robinet, P.; Berisha, S.; Smith, J.D. Soat1 mediates the mouse strain effects on cholesterol loading-induced endoplasmic reticulum stress and CHOP expression in macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158825. [Google Scholar] [CrossRef] [PubMed]
- Li, B.L.; Li, X.L.; Duan, Z.J.; Lee, O.; Lin, S.; Ma, Z.M.; Chang, C.C.; Yang, X.Y.; Park, J.P.; Mohandas, T.K.; et al. Human acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) gene organization and evidence that the 4.3-kilobase ACAT-1 mRNA is produced from two different chromosomes. J. Biol. Chem. 1999, 274, 11060–11071. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.-J.; Chen, J.; Zhao, X.-N.; Xu, J.-J.; Guo, D.-Q.; Lu, M.; Zhu, M.; Xiong, Y.; Li, Q.; Chang, C.C.; et al. Production of ACAT1 56-kDa isoform in human cells via trans-splicing involving the ampicillin resistance gene. Cell Res. 2013, 23, 1007–1024. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Li, C.; Zuo, Z.; Huang, C.; Cheng, H.; Zhou, R. Evolutionary Insights into RNA trans-Splicing in Vertebrates. Genome Biol. Evol. 2016, 8, 562–577. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yuan, C.; Chen, L.; Lei, M.; Zellmer, L.; Huang, H.; Liao, D.J. Transcriptional-Readthrough RNAs Reflect the Phenomenon of “A Gene Contains Gene(s)” or “Gene(s) within a Gene” in the Human Genome, and Thus Are Not Chimeric RNAs. Genes 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P.; Ntzani, E.E.; Trikalinos, T.A.; Contopoulos-Ioannidis, D.G. Replication validity of genetic association studies. Nat. Genet. 2001, 29, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Takata, K.; Katsuren, K.; Fukuyama, S. The influence of the acyl-CoA:cholesterol acyltransferase-1 gene (-77G-->A) polymorphisms on plasma lipid and apolipoprotein levels in normolipidemic and hyperlipidemic subjects. Biochim. Biophys. Acta 2004, 1682, 56–62. [Google Scholar] [CrossRef]
- Chen, S.N.; Cilingiroglu, M.; Todd, J.; Lombardi, R.; Willerson, J.T.; Gotto, A.M.J.; Ballantyne, C.M.; Marian, A.J. Candidate genetic analysis of plasma high-density lipoprotein-cholesterol and severity of coronary atherosclerosis. BMC Med. Genet. 2009, 10, 111. [Google Scholar] [CrossRef]
- Wu, D.-F.; Yin, R.-X.; Aung, L.H.H.; Hu, X.-J.; Cao, X.-L.; Miao, L.; Li, Q.; Yan, T.-T.; Wu, J.-Z.; Pan, S.-L. Polymorphism of rs1044925 in the acyl-CoA:cholesterol acyltransferase-1 gene and serum lipid levels in the Guangxi Bai Ku Yao and Han populations. Lipids Health Dis. 2010, 9, 139. [Google Scholar] [CrossRef]
- Wu, D.-F.; Yin, R.-X.; Aung, L.H.H.; Li, Q.; Yan, T.-T.; Zeng, X.-N.; Huang, K.-K.; Huang, P.; Wu, J.-Z.; Pan, S.-L. Sex-specific association of ACAT-1 rs1044925 SNP and serum lipid levels in the hypercholesterolemic subjects. Lipids Health Dis. 2012, 11, 9. [Google Scholar] [CrossRef]
- Wu, D.-F.; Yin, R.-X.; Cao, X.-L.; Chen, W.-X. Association between single nucleotide polymorphism rs1044925 and the risk of coronary artery disease and ischemic stroke. Int. J. Mol. Sci. 2014, 15, 3546–3559. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Wang, Y.-H.; Ma, Y.-T.; Fu, Z.-Y.; Yang, Y.-N.; Ma, X.; Li, X.-M.; Adi, D.; Liu, F.; Chen, B.-D. ACAT-1 gene polymorphism is associated with increased susceptibility to coronary artery disease in Chinese Han population: A case-control study. Oncotarget 2017, 8, 89055–89063. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Maitusong, B.; Ma, Y.-T.; Fu, Z.-Y.; Yang, Y.-N.; Ma, X.; Li, X.-M.; Liu, F.; Chen, B.-D. Acyl-CoA: Cholesterol acyltransferases-2 gene polymorphism is associated with increased susceptibility to coronary artery disease in Uygur population in Xinjiang, China. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.B.; Padovani, C.R.; de Oliveira Junior, L.R.; Latini, A.C.P.; Kurokawa, C.S.; Pereira, P.C.M.; Dos Santos, R.M. ACAT-1 gene rs1044925 SNP and its relation with different clinical forms of chronic Chagas disease. Parasitol. Res. 2019, 118, 2343–2351. [Google Scholar] [CrossRef]
- Huang, L.-H.; Nishi, K.; Li, S.; Ho, T.; Dong, R.; Chang, C.C.Y.; Chang, T.-Y. Acyl-coenzyme A:cholesterol acyltransferase 1—significance of single-nucleotide polymorphism at residue 526 and the role of Pro347 near the fifth transmembrane domain. FEBS J. 2014, 281, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Klos, K.L.E.; Sing, C.F.; Boerwinkle, E.; Hamon, S.C.; Rea, T.J.; Clark, A.; Fornage, M.; Hixson, J.E. Consistent effects of genes involved in reverse cholesterol transport on plasma lipid and apolipoprotein levels in CARDIA participants. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, A.C.; Braund, P.S.; Stylianou, I.M.; Khera, A.V.; Nelson, C.P.; Wolfe, M.L.; Derohannessian, S.L.; Keating, B.J.; Qu, L.; He, J.; et al. Dense genotyping of candidate gene loci identifies variants associated with high-density lipoprotein cholesterol. Circ. Cardiovasc. Genet. 2011, 4, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Wollmer, M.A.; Streffer, J.R.; Tsolaki, M.; Grimaldi, L.M.E.; Lütjohann, D.; Thal, D.; von Bergmann, K.; Nitsch, R.M.; Hock, C.; Papassotiropoulos, A. Genetic association of acyl-coenzyme A: Cholesterol acyltransferase with cerebrospinal fluid cholesterol levels, brain amyloid load, and risk for Alzheimer’s disease. Mol. Psychiatry 2003, 8, 635–638. [Google Scholar] [CrossRef][Green Version]
- Papassotiropoulos, A.; Wollmer, M.A.; Tsolaki, M.; Brunner, F.; Molyva, D.; Lütjohann, D.; Nitsch, R.M.; Hock, C. A cluster of cholesterol-related genes confers susceptibility for Alzheimer’s disease. J. Clin. Psychiatry 2005, 66, 940–947. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Greco, C.; Kovacs, D.M. Knockdown of ACAT-1 reduces amyloidogenic processing of APP. FEBS Lett. 2007, 581, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Kovacs, D.M. ACAT inhibition and amyloid beta reduction. Biochim. Biophys. Acta 2010, 1801, 960–965. [Google Scholar] [CrossRef]
- Murphy, S.R.; Chang, C.C.; Dogbevia, G.; Bryleva, E.Y.; Bowen, Z.; Hasan, M.T.; Chang, T.-Y. Acat1 knockdown gene therapy decreases amyloid-β in a mouse model of Alzheimer’s disease. Mol. Ther. 2013, 21, 1497–1506. [Google Scholar] [CrossRef]
- Bryleva, E.Y.; Rogers, M.A.; Chang, C.C.Y.; Buen, F.; Harris, B.T.; Rousselet, E.; Seidah, N.G.; Oddo, S.; LaFerla, F.M.; Spencer, T.A.; et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. USA 2010, 107, 3081–3086. [Google Scholar] [CrossRef]
- Naelitz, B.D.; Sharifi, N. Through the Looking-Glass: Reevaluating DHEA Metabolism Through HSD3B1 Genetics. Trends Endocrinol. Metab. 2020, 31, 680–690. [Google Scholar] [CrossRef]
- Jansen, P.R.; Watanabe, K.; Stringer, S.; Skene, N.; Bryois, J.; Hammerschlag, A.R.; de Leeuw, C.A.; Benjamins, J.S.; Muñoz-Manchado, A.B.; Nagel, M.; et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet. 2019, 51, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Ruth, K.S.; Day, F.R.; Tyrrell, J.; Thompson, D.J.; Wood, A.R.; Mahajan, A.; Beaumont, R.N.; Wittemans, L.; Martin, S.; Busch, A.S.; et al. Using human genetics to understand the disease impacts of testosterone in men and women. Nat. Med. 2020, 26, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, J.; Ibrahim, J.G.; Luo, T.; Santelli, R.C.; Li, Y.; Li, T.; Shan, Y.; Zhu, Z.; Zhou, F.; et al. Large-scale GWAS reveals genetic architecture of brain white matter microstructure and genetic overlap with cognitive and mental health traits (n = 17,706). Mol. Psychiatry 2019, 1–13. [Google Scholar] [CrossRef]
- Yan, Q.; Ding, Y.; Liu, Y.; Sun, T.; Fritsche, L.G.; Clemons, T.; Ratnapriya, R.; Klein, M.L.; Cook, R.J.; Liu, Y.; et al. Genome-wide analysis of disease progression in age-related macular degeneration. Hum. Mol. Genet. 2018, 27, 929–940. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Name | Animal | Plasma Cholesterol | Atherosclerotic Lesion Effect | Other Effects | Reference |
|---|---|---|---|---|---|
| F-1394 | Rats | ↓ | N.D. | ↓ cholesterol absorption | [31] |
| Apoe−/− ldlr−/− mice | N.S. | ↓ | [32] | ||
| Rabbits (balloon injury) | N.S. | N.D. | ↓ neointimal thickening | [33] | |
| Apoe−/− mice | ↓ | ↓ | [34] | ||
| Apoe−/− mice | N.S. | ↓ progression | ↓ lesion tissue factor | [35] | |
| Apoe−/− mice | N.S. | N.S. | [36] | ||
| Apoe−/− and human APO A1-transgenic mice | N.S. | N.S. | [36] | ||
| CI-1011 (avasimibe) | Male monkeys | ↓ | N.D. | ↓ Lp(a) | [37] |
| Miniature pigs | ↓ | N.D. | [38] | ||
| Male rabbits | N.S. | ↓ progression | ↓ lesion MMP expression | [39] | |
| Beagle dogs | N.D. | N.D. | ↑ Emesis, saliva, hepatic toxicity, | [40] | |
| ApoE *3-Leiden mice | ↓ | ↓ | [41] | ||
| Miniature pigs | ↓ | N.D. | [42] | ||
| Apoe−/− mice | ↓ | ↓ progression | [43] | ||
| Rabbits | N.D. | ↓ progression | [44] | ||
| CS-505 (pactimibe sulfate) | Apoe−/− mice | ↓ | ↓ progression | ↓ MMP expression | [43] |
| Rabbits | N.S. | N.S. | ↑ lesion fibers and smooth muscle cells | [45] | |
| Hamsters | ↓ | ↓ | [46] | ||
| Hamsters | ↓ | N.D. | ↓ hepatic lipids; ↓ lipid absorption; ↑ fecal lipid excretion | [47] | |
| Monkeys | ↓ | N.D. | [47] | ||
| Diabetic rats | N.D. | N.D. | ↓ postprandial fat loading | [47] | |
| Apoe−/− mice | ↓ | ↓ | [48] | ||
| CI-976 | Rabbits | N.S. | ↓ progression | [49] | |
| Hamsters | ↓ | N.D. | [50] | ||
| Rats | ↓ | N.D. | ↓ liver CE | [51] | |
| Micropigs | N.S. | N.D. | ↓ liver CE | [52] | |
| Endogenous hypercholesterolemia rabbits | ↓ | N.D. | [53] | ||
| Rabbits | ↓ | ↓ | [54] | ||
| Nephrotic syndrome (NS) rats | ↓ | N.D. | restore LDL and HDL receptors | [55] | |
| K-604 (ACAT-1 selective) | Hamsters | ↓ (at high dose) | ↓ | [56] | |
| Apoe−/− mice | ↓ (at high dose) | N.S. | ↑ lesion collagen | [48] | |
| PD 132301-2 | Rats | ↓ | N.D. | [57] | |
| Guinea pigs | ↓ | N.D. | [57] | ||
| Rabbits | ↓ | N.D. | [57] | ||
| CL 277,082 | Rats | ↓ | N.D. | [57] | |
| Guinea pigs | ↓ (at high dose) | N.D. | [57] | ||
| Rabbits | ↓ | N.D. | [57] | ||
| Rats | ↓ | N.D. | [51] |
| Gene | Variant | p-Value | Reported Trait | Reference |
|---|---|---|---|---|
| SOAT1 | rs13306728 | 6 × 10−14 | Morningness | [102] |
| rs2152318 | 2 × 10−27 | Bioavailable testosterone levels | [103] | |
| rs2248979 | 2 × 10−12 | Bioavailable testosterone levels | [103] | |
| rs569421885 | 1 × 10−62 | Total testosterone levels | [103] | |
| rs569421885 | 3 × 10−37 | Total testosterone levels | [103] | |
| rs2816177 | 7 × 10−9 | Type 2 diabetes | [104] | |
| rs67563284 | 2 × 10−8 | White matter microstructure | [105] | |
| SOAT2 | rs11170417 | 7 × 10−6 | Age-related macular degeneration progression | [106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hai, Q.; Smith, J.D. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era. Metabolites 2021, 11, 543. https://doi.org/10.3390/metabo11080543
Hai Q, Smith JD. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era. Metabolites. 2021; 11(8):543. https://doi.org/10.3390/metabo11080543
Chicago/Turabian StyleHai, Qimin, and Jonathan D. Smith. 2021. "Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era" Metabolites 11, no. 8: 543. https://doi.org/10.3390/metabo11080543
APA StyleHai, Q., & Smith, J. D. (2021). Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era. Metabolites, 11(8), 543. https://doi.org/10.3390/metabo11080543