
TrpNet: Understanding Tryptophan Metabolism across Gut Microbiome



Abstract
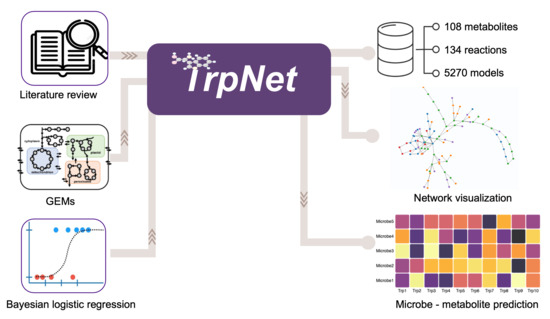
:
1. Introduction
2. Results
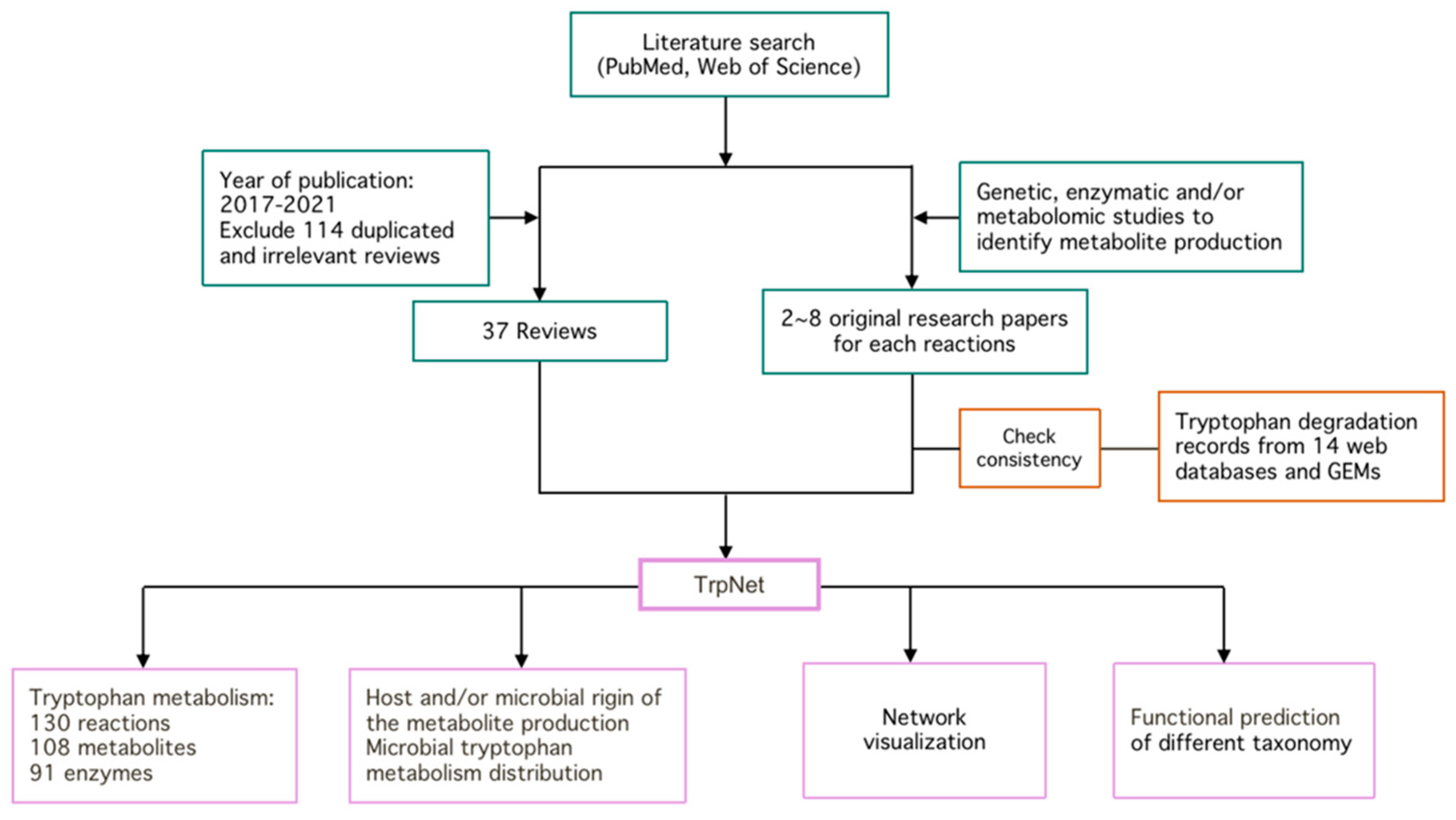
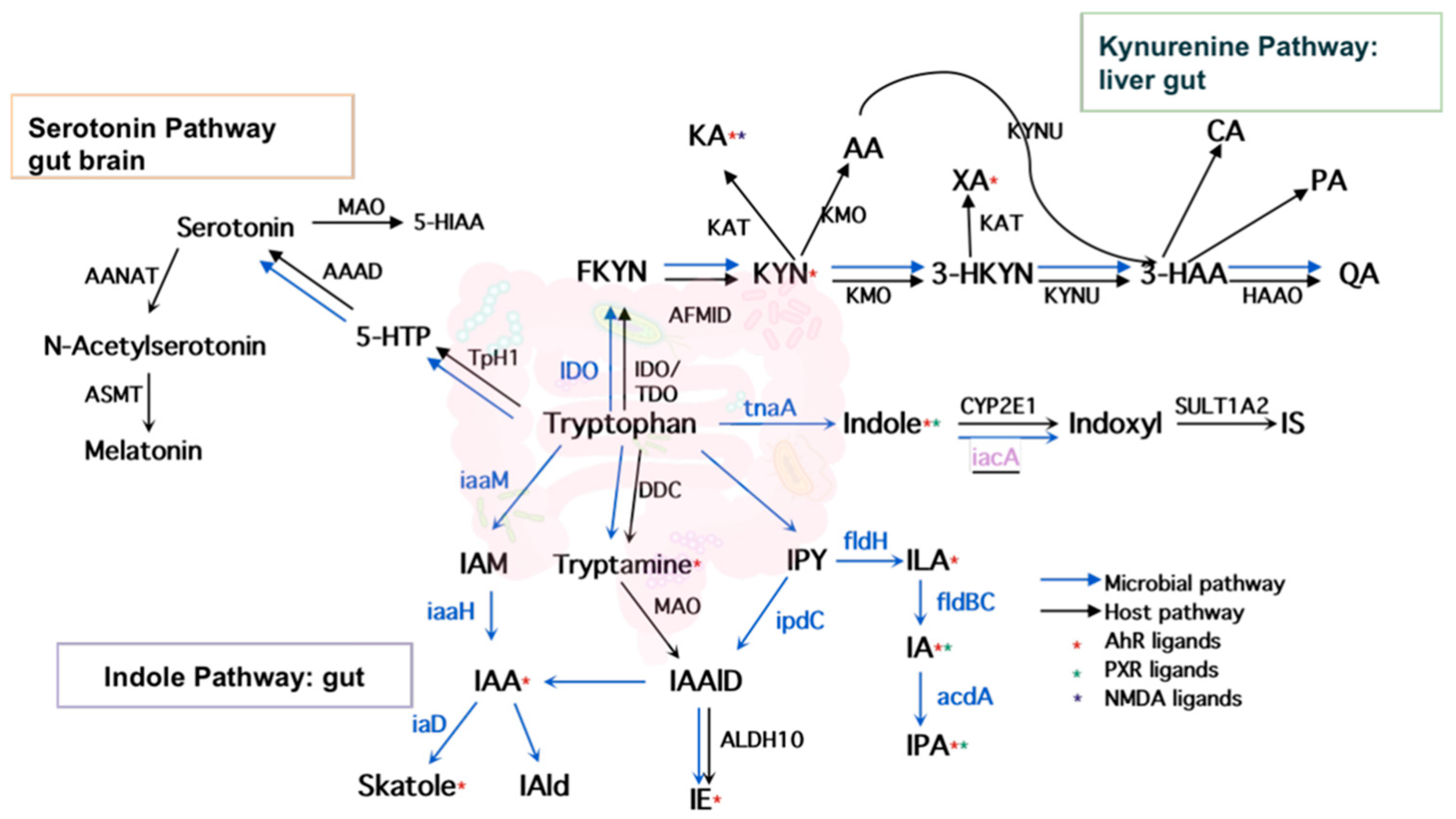
2.1. Literature Search and Intestinal Tryptophan Metabolism
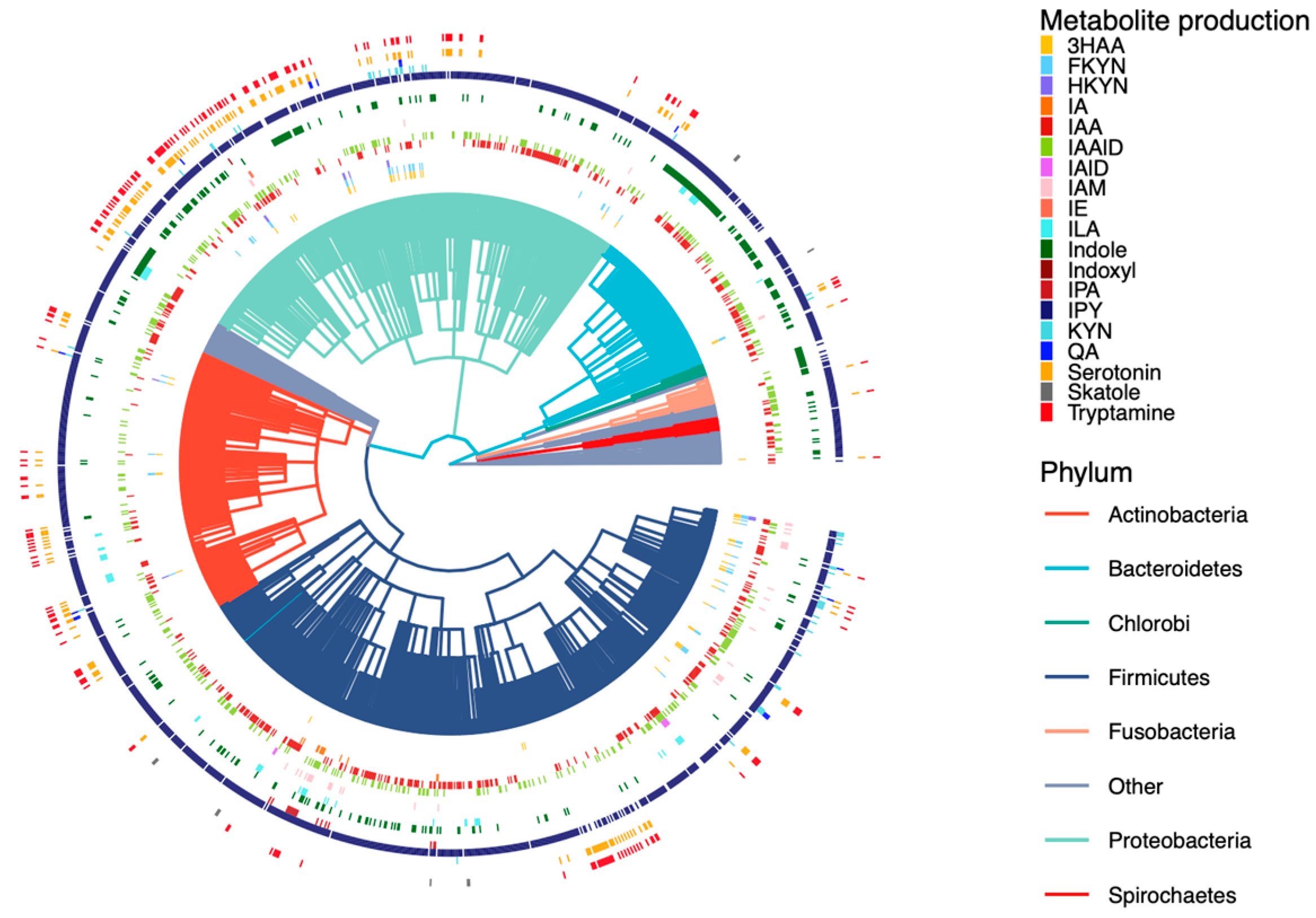
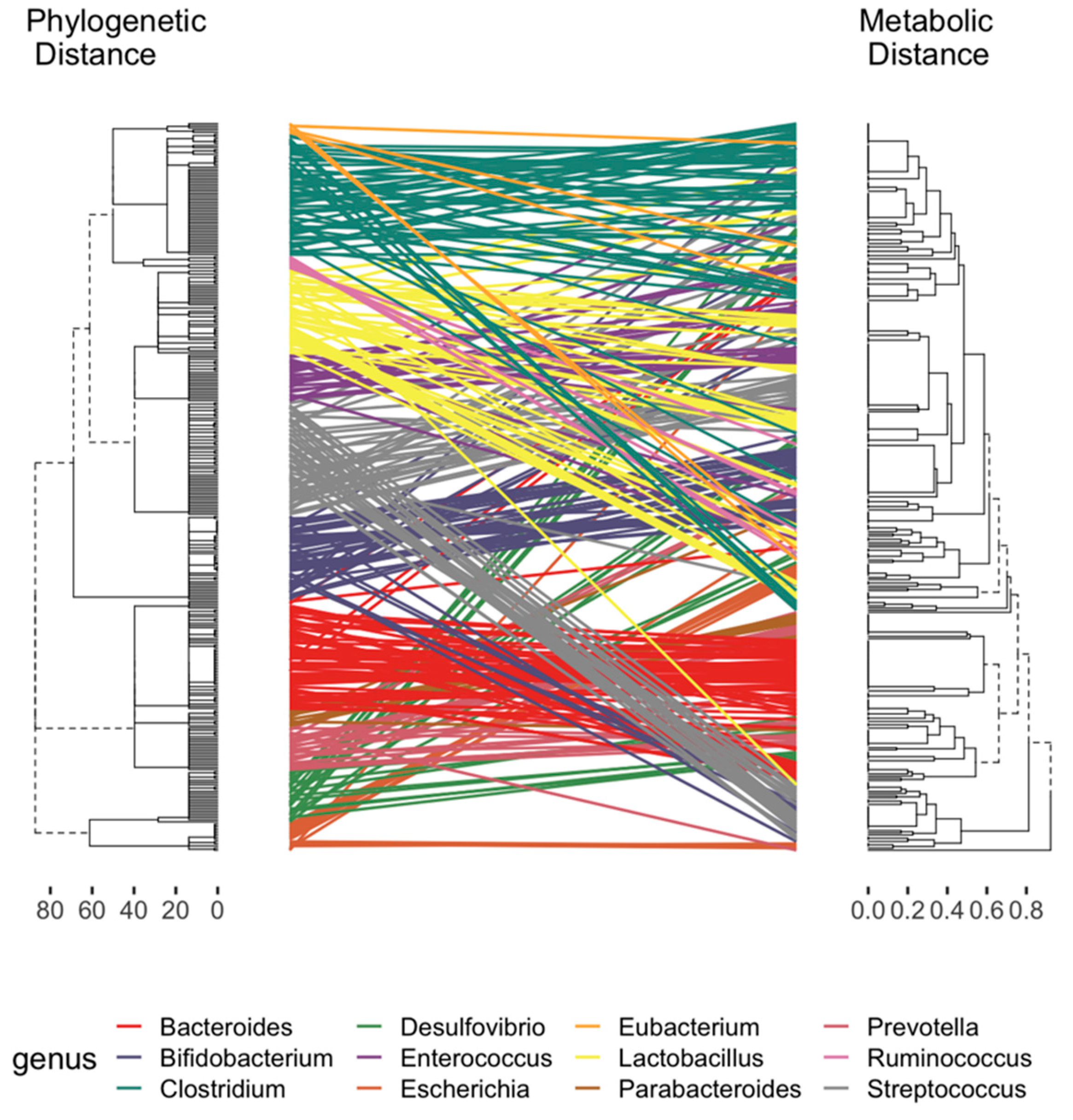
2.2. Curation of Genome-Scale Metabolic Models
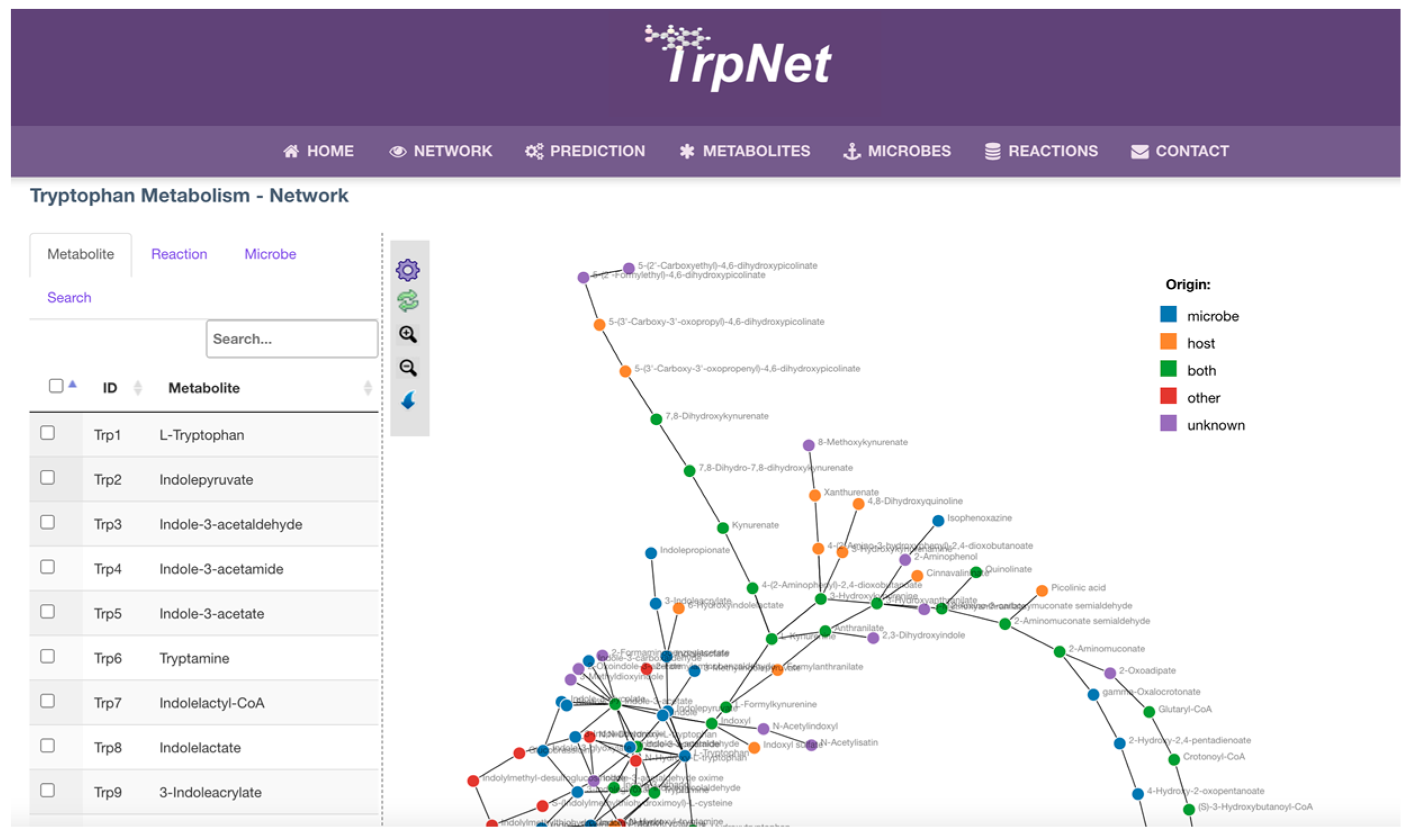
2.3. Development of a Database for Tryptophan Metabolism and Functional Prediction
2.4. Case Studies
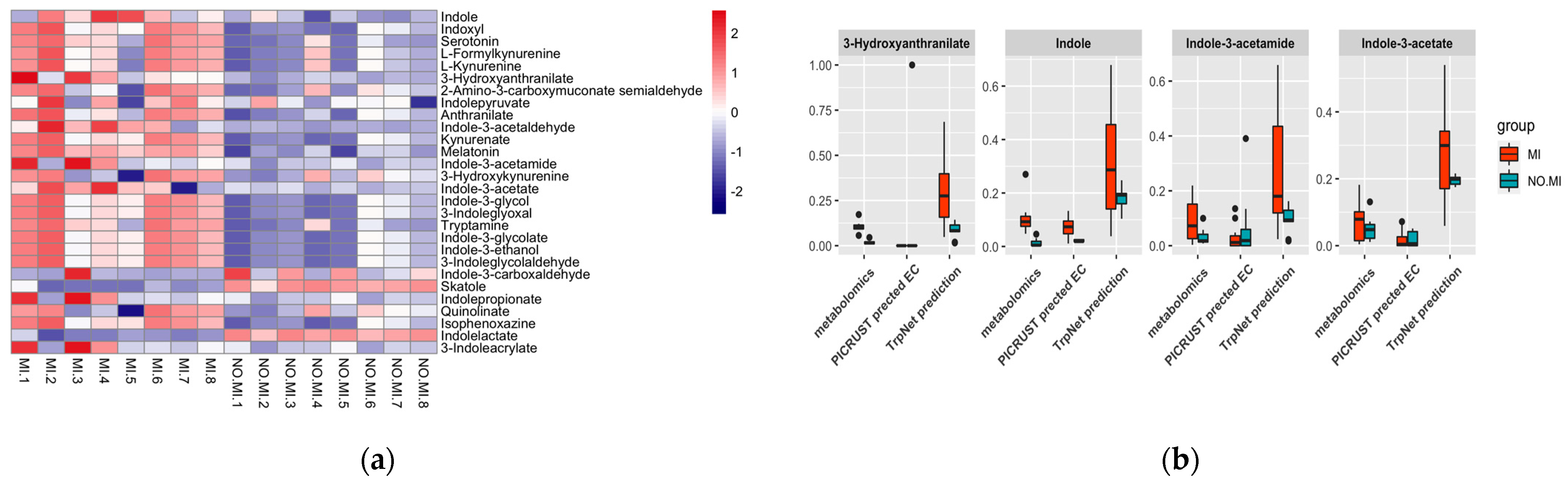
2.4.1. Myocardial Infarct (MI) Case Study
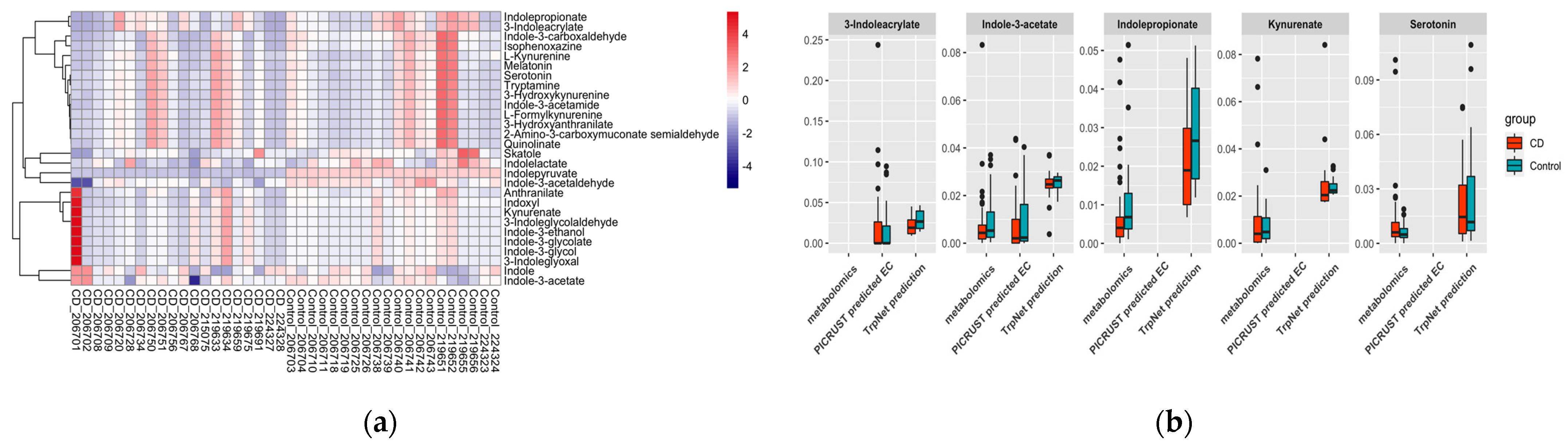
2.4.2. IBD Case Study
3. Discussion
4. Materials and Methods
4.1. Literature Review
4.2. GEMs Collection
4.3. TrpNet Implementation
4.4. Sample Collection for MI Case Study
4.5. Sample Collection for IBD Case Study
4.6. Logistic Regression Model for Predicting Metabolite Profiles
- Different taxonomy levels and their combinations were evaluated for their predictive values. Models were ranked by Akaike information criterion (AIC). The genus level combined with the host type was selected as the best predictor;
- The models were further optimized by Bayesian logistic regression coupled with a fast Pareto smoothed leave-one-out cross-validation for the penalized likelihood estimation [67]. These models capture the metabolite production potential (PM, G) for the underlying metabolite (M) of interest in every genus (G) for a given host type;
- The predicted probability (PM,G) was multiplied by the genus abundance table obtained from 16S rRNA sequencing data to compute the accumulated production potential for each metabolite of interest for each sample;
- The results of all samples were normalized by total sum scaling to be comparable with metabolomics data.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dethlefsen, L.; McFall-Ngai, M.; Relman, D.A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 2007, 449, 811–818. [Google Scholar] [CrossRef]
- Chassaing, B.; Darfeuille-Michaud, A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Mendonca, L.; Dhariwal, A.; Fontes, G.; Menzies, D.; Xia, J.; Divangahi, M.; King, I.L. Intestinal dysbiosis compromises alveolar macrophage immunity to Mycobacterium tuberculosis. Mucosal. Immunol. 2019, 12, 772–783. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Sampson, T.R.; Mazmanian, S.K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 2015, 17, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-Modulated Metabolites at the Interface of Host Immunity. J. Immunol. 2017, 198, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Hirota, K.; Duarte, J.; Veldhoen, M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin. Immunol. 2011, 23, 99–105. [Google Scholar] [CrossRef]
- Jaichander, P.; Selvarajan, K.; Garelnabi, M.; Parthasarathy, S. Induction of paraoxonase 1 and apolipoprotein A-I gene expression by aspirin. J. Lipid Res. 2008, 49, 2142–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Qi, Q.; Li, J.; Yu, B.; Moon, J.Y.; Chai, J.C.; Merino, J.; Hu, J.; Ruiz-Canela, M.; Rebholz, C.; Wang, Z.; et al. Host and gut microbial tryptophan metabolism and type 2 diabetes: An integrative analysis of host genetics, diet, gut microbiome and circulating metabolites in cohort studies. Gut 2021. [Google Scholar] [CrossRef]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Jewison, T.; Su, Y.; Disfany, F.M.; Liang, Y.; Knox, C.; Maciejewski, A.; Poelzer, J.; Huynh, J.; Zhou, Y.; Arndt, D.; et al. SMPDB 2.0: Big improvements to the Small Molecule Pathway Database. Nucleic Acids Res 2014, 42, D478–D484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting communities. Nucleic Acids Res 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef]
- Han, S.; Van Treuren, W.; Fischer, C.R.; Merrill, B.D.; DeFelice, B.C.; Sanchez, J.M.; Higginbottom, S.K.; Guthrie, L.; Fall, L.A.; Dodd, D.; et al. A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature 2021, 595, 415–420. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2020, 49, D575–D588. [Google Scholar] [CrossRef]
- Bauer, E.; Laczny, C.C.; Magnusdottir, S.; Wilmes, P.; Thiele, I. Phenotypic differentiation of gastrointestinal microbes is reflected in their encoded metabolic repertoires. Microbiome 2015, 3, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Haleem, A.M.; Hefzi, H.; Mineta, K.; Gao, X.; Gojobori, T.; Palsson, B.O.; Lewis, N.E.; Jamshidi, N. Functional interrogation of Plasmodium genus metabolism identifies species- and stage-specific differences in nutrient essentiality and drug targeting. PLoS Comput. Biol. 2018, 14, e1005895. [Google Scholar] [CrossRef]
- Zhang, C.; Shao, H.; Li, D.; Xiao, N.; Tan, Z. Role of tryptophan-metabolizing microbiota in mice diarrhea caused by Folium sennae extracts. BMC Microbiol. 2020, 20, 185. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Özoğul, F.; Kuley, E.; Özoğul, Y.; Özoğul, İ. The Function of Lactic Acid Bacteria on Biogenic Amines Production by Food-Borne Pathogens in Arginine Decarboxylase Broth. Food Sci. Technol. Res. 2012, 18, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Özoğul, F. Production of biogenic amines by Morganella morganii, Klebsiella pneumoniae and Hafnia alvei using a rapid HPLC method. Eur. Food Res. Technol. 2004, 219, 465–469. [Google Scholar] [CrossRef]
- Kitahama, K.; Ikemoto, K.; Jouvet, A.; Nagatsu, I.; Sakamoto, N.; Pearson, J. Aromatic L-amino acid decarboxylase- and tyrosine hydroxylase-immunohistochemistry in the adult human hypothalamus. J. Chem. Neuroanat. 1998, 16, 43–55. [Google Scholar] [CrossRef]
- Colabroy, K.L.; Begley, T.P. Tryptophan catabolism: Identification and characterization of a new degradative pathway. J. Bacteriol. 2005, 187, 7866–7869. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hou, Y.; Wang, G.; Zheng, X.; Hao, H. Gut Microbial Metabolites of Aromatic Amino Acids as Signals in Host-Microbe Interplay. Trends Endocrinol. Metab. 2020, 31, 818–834. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J. Systems Biology of Metabolism. Annu. Rev. Biochem. 2017, 86, 245–275. [Google Scholar] [CrossRef]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019, 20, 158. [Google Scholar] [CrossRef] [Green Version]
- Forster, S.C.; Kumar, N.; Anonye, B.O.; Almeida, A.; Viciani, E.; Stares, M.D.; Dunn, M.; Mkandawire, T.T.; Zhu, A.; Shao, Y.; et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 2019, 37, 186–192. [Google Scholar] [CrossRef]
- Zou, Y.; Xue, W.; Luo, G.; Deng, Z.; Qin, P.; Guo, R.; Sun, H.; Xia, Y.; Liang, S.; Dai, Y.; et al. 1520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019, 37, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Pukall, R.; Abt, B.; Foesel, B.U.; Meier-Kolthoff, J.P.; Kumar, N.; Bresciani, A.; Martínez, I.; Just, S.; Ziegler, C.; et al. The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat. Microbiol. 2016, 1, 16131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhou, N.; Du, M.-X.; Sun, Y.-T.; Wang, K.; Wang, Y.-J.; Li, D.-H.; Yu, H.-Y.; Song, Y.; Bai, B.-B.; et al. The Mouse Gut Microbial Biobank expands the coverage of cultured bacteria. Nat. Commun. 2020, 11, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmes, P.; Bowen, B.P.; Thomas, B.C.; Mueller, R.S.; Denef, V.J.; VerBerkmoes, N.C.; Hettich, R.L.; Northen, T.R.; Banfield, J.F. Metabolome-proteome differentiation coupled to microbial divergence. MBio 2010, 1, e00246-10. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef]
- Ramadoss, P.; Perdew, G.H. Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol. Pharmacol. 2004, 66, 129–136. [Google Scholar] [CrossRef]
- Dong, M.; Li, L.; Chen, M.; Kusalik, A.; Xu, W. Predictive analysis methods for human microbiome data with application to Parkinson’s disease. PLoS ONE 2020, 15, e0237779. [Google Scholar] [CrossRef]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Loncar Turukalo, T.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef]
- Xia, F.; Chen, J.; Fung, W.K.; Li, H. A logistic normal multinomial regression model for microbiome compositional data analysis. Biometrics 2013, 69, 1053–1063. [Google Scholar] [CrossRef]
- Mangge, H.; Stelzer, I.; Reininghaus, E.Z.; Weghuber, D.; Postolache, T.T.; Fuchs, D. Disturbed tryptophan metabolism in cardiovascular disease. Curr. Med. Chem. 2014, 21, 1931–1937. [Google Scholar] [CrossRef]
- Millett, E.R.C.; Peters, S.A.E.; Woodward, M. Sex differences in risk factors for myocardial infarction: Cohort study of UK Biobank participants. BMJ 2018, 363, k4247. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Shen, X.; Wu, S.; Liang, L.; Chen, S.; Contrepois, K.; Zhu, Z.J.; Snyder, M. metID: A R package for automatable compound annotation for LC-MS-based data. Bioinformatics 2021. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Kerley-Hamilton, J.S.; Trask, H.W.; Ridley, C.J.; Dufour, E.; Lesseur, C.; Ringelberg, C.S.; Moodie, K.L.; Shipman, S.L.; Korc, M.; Gui, J.; et al. Inherent and benzo[a]pyrene-induced differential aryl hydrocarbon receptor signaling greatly affects life span, atherosclerosis, cardiac gene expression, and body and heart growth in mice. Toxicol. Sci. 2012, 126, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Shui, X.; He, Y.; Xue, Y.; Li, J.; Li, G.; Lei, W.; Chen, C. AhR expression and polymorphisms are associated with risk of coronary arterial disease in Chinese population. Sci. Rep. 2015, 5, 8022. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Nishimura, N.; Kuo, V.; Fiehn, O.; Shahbaz, S.; Van Winkle, L.; Matsumura, F.; Vogel, C.F. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E-/- mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Manzella, C.R.; Jayawardena, D.; Pagani, W.; Li, Y.; Alrefai, W.A.; Bauer, J.; Jung, B.; Weber, C.R.; Gill, R.K. Serum Serotonin Differentiates Between Disease Activity States in Crohn’s Patients. Inflamm. Bowel Dis. 2020, 26, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.B.; Kasneci, A.; Bolt, A.M.; Di Lalla, V.; Di Iorio, M.R.; Raad, M.; Mann, K.K.; Chalifour, L.E. Chronic Exposure to Bisphenol a Reduces Successful Cardiac Remodeling After an Experimental Myocardial Infarction in Male C57bl/6n Mice. Toxicol. Sci. 2015, 146, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Kasneci, A.; Lee, J.S.; Yun, T.J.; Shang, J.; Lampen, S.; Gomolin, T.; Cheong, C.C.; Chalifour, L.E. From the Cover: Lifelong Exposure of C57bl/6n Male Mice to Bisphenol A or Bisphenol S Reduces Recovery from a Myocardial Infarction. Toxicol. Sci. 2017, 159, 189–202. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.E.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Integrative, H.M.P.R.N.C. The Integrative Human Microbiome Project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Vehtari, A.; Gelman, A.; Gabry, J. Practical Bayesian model evaluation using leave-one-out cross-validation and WAIC. Stat Comput. 2017, 27, 1413–1432. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predictors | IA | Indole | IAAlD | IAM | IAA | ILA | IPA | Tryptamine |
|---|---|---|---|---|---|---|---|---|
| Bacteroides | 0.8786 | 310.4118 | 1.5256 | 0.5621 | 2.9424 | 69.0048 | 0.8515 | 0.1484 |
| Bifidobacterium | 0.8712 | 0.0421 | 0.4879 | 0.6081 | 1.0597 | 103.1476 | 0.8393 | 8.5582 |
| Clostridium | 413.0681 | 2.2526 | 1.6328 | 106.6308 | 0.8401 | 89.0063 | 638.3164 | 3.473 |
| Desulfovibrio | 0.9738 | 1.5226 | 1.2451 | 0.8139 | 14.9215 | 0.8931 | 0.9676 | 0.3856 |
| Enterococcus | 0.9225 | 2.1667 | 0.8861 | 0.7017 | 0.0392 | 0.6446 | 0.9017 | 1.0872 |
| Escherichia | 0.936 | 241.1231 | 0.087 | 0.7997 | 3.9424 | 226.036 | 0.9161 | 9.0402 |
| Eubacterium | 0.9783 | 3.7121 | 0.622 | 0.8615 | 16.7253 | 0.8792 | 0.9723 | 0.4817 |
| Lactobacillus | 0.8536 | 0.0324 | 1.9794 | 0.5087 | 1.765 | 36.9424 | 0.8227 | 0.4338 |
| Mouse.gut | 2.5942 | 2.2931 | 1.1424 | 0.937 | 1.8319 | 6.9211 | 2.9119 | 0.4471 |
| Parabacteroides | 0.9668 | 0.1841 | 0.9344 | 0.8384 | 1.2605 | 13.8291 | 0.9569 | 0.4585 |
| Prevotella | 0.9145 | 1.6855 | 0.7029 | 0.5947 | 0.473 | 0.7286 | 0.8969 | 0.1589 |
| Ruminococcus | 0.9718 | 0.7449 | 0.4029 | 0.8035 | 7.2895 | 0.8861 | 0.9651 | 0.3699 |
| Streptococcus | 0.8959 | 0.6288 | 0.7287 | 0.5533 | 0.527 | 0.6756 | 0.8749 | 19.0245 |
| Predictors | IA | Indole | IAAlD | IAM | IAA | ILA | IPA | Tryptamine |
|---|---|---|---|---|---|---|---|---|
| Bacteroides | 0.8855 | 1595.5832 | 1.4595 | 0.5265 | 2.6489 | 79.5618 | 0.8575 | 0.1214 |
| Bifidobacterium | 0.9025 | 0.0371 | 0.7658 | 0.5674 | 0.7158 | 175.3057 | 0.8781 | 5.5164 |
| Clostridium | 414.0254 | 1.8606 | 1.2366 | 91.4966 | 1.6268 | 81.5643 | 606.5603 | 2.8663 |
| Desulfovibrio | 0.9683 | 1.55 | 0.6004 | 0.79 | 47.2592 | 0.8512 | 0.9593 | 0.3552 |
| Enterococcus | 0.9478 | 2.561 | 1.2673 | 0.7037 | 0.0322 | 0.7858 | 0.9333 | 0.9879 |
| Escherichia | 0.9639 | 318.856 | 0.081. | 0.7677 | 4.0763 | 607.7244 | 0.9531 | 4.6206 |
| Eubacterium | 0.981 | 2.0208 | 1.2466 | 0.8559 | 10.5365 | 0.9035 | 0.9753 | 0.463 |
| Human.gut | 21.3204 | 1.4413 | 0.7778 | 342.7406 | 2.1685 | 20.4853 | 37.332 | 1876.3277 |
| Lactobacillus | 0.8757 | 0.1256 | 2.4492 | 0.5055 | 1.5343 | 52.7602 | 0.8462 | 0.412 |
| Parabacteroides | 0.976 | 0.1964 | 1.3015 | 0.8271 | 1.681 | 28.2969 | 0.9687 | 0.4121 |
| Prevotella | 0.9479 | 3.5273 | 2.0026 | 0.7035 | 1.294 | 0.7879 | 0.9332 | 0.2527 |
| Ruminococcus | 0.9663 | 0.6021 | 0.3477 | 0.7784 | 4.9819 | 0.8487 | 0.9562 | 0.3397 |
| Streptococcus | 0.8871 | 0.451 | 0.7435 | 0.5304 | 0.3467 | 0.6357 | 0.8596 | 18.3925 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Chong, J.; Shen, S.; Chammas, J.-B.; Chalifour, L.; Xia, J. TrpNet: Understanding Tryptophan Metabolism across Gut Microbiome. Metabolites 2022, 12, 10. https://doi.org/10.3390/metabo12010010
Lu Y, Chong J, Shen S, Chammas J-B, Chalifour L, Xia J. TrpNet: Understanding Tryptophan Metabolism across Gut Microbiome. Metabolites. 2022; 12(1):10. https://doi.org/10.3390/metabo12010010
Chicago/Turabian StyleLu, Yao, Jasmine Chong, Shiqian Shen, Joey-Bahige Chammas, Lorraine Chalifour, and Jianguo Xia. 2022. "TrpNet: Understanding Tryptophan Metabolism across Gut Microbiome" Metabolites 12, no. 1: 10. https://doi.org/10.3390/metabo12010010
APA StyleLu, Y., Chong, J., Shen, S., Chammas, J.-B., Chalifour, L., & Xia, J. (2022). TrpNet: Understanding Tryptophan Metabolism across Gut Microbiome. Metabolites, 12(1), 10. https://doi.org/10.3390/metabo12010010