
A Gas Chromatography Mass Spectrometry-Based Method for the Quantification of Short Chain Fatty Acids

 , and
, and
Abstract
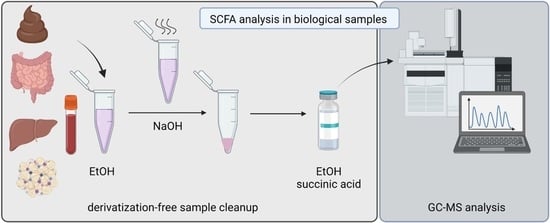
:
1. Introduction
2. Results and Discussion
2.1. GC-MS Method
2.2. Calibration Curve, Limit of Quantification, Carry Over, Accuracy and Precision
2.3. Sample Extraction and Recovery
2.4. Matrix Effects
2.5. Method Application
3. Materials and Methods
3.1. Chemicals
3.2. Standard Solutions and Calibration
3.3. Sample Preparation
3.4. GC. MS Analysis
3.5. Linearity
3.6. Limit of Dectection (LOD) and Quantification (LOQ)
3.7. Carry Over
3.8. Recovery Assay
3.9. Standard-Addition
3.10. Effects of Matrix Effects
3.11. Reproducibilty
3.12. Experimental Animals
3.13. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | m/z (TI, CI1, CI2) | RT | Used for | Carry Over (% LOQ) |
|---|---|---|---|---|
| Acetic acid d4 | 46. 63 | 5.4 | Acetic acid | 0.15 |
| Propionic acid d6 | 30. 46; 79 | 6.5 | Propionic acid | 0.12 |
| Butyric acid d7 | 63, 77 | 7.5 | Butyric acid, Isovaleric acid Valeric acid 4-Methylvaleric acid Hexanoic acid | 0.13 |
| Analyte | L1 | L2 | L3 | |||||||||
| C (µg/mL) | BCC (µg/mL) | RSD (%) | Accuracy RE (%) | C (µg/mL) | BCC (µg/mL) | RSD (%) | Accuracy RE (%) | C (µg/mL) | BCC (µg/mL) | RSD (%) | Accuracy RE (%) | |
| Acetic acid | 10 | 9.94 | 2.49 | 0.58 | 25 | 26.27 | 2.65 | 5.07 | 50 | 50.45 | 2.38 | 0.89 |
| Propionic acid | 5 | 7.41 | 7.41 | 48.28 | 12.5 | 13.62 | 6.14 | 8.98 | 25 | 24.85 | 3.48 | 0.59 |
| Butyric acid | 10 | 0.65 | 5.95 | 93.48 | 25 | 19.73 | 3.29 | 21.07 | 50 | 47.86 | 4.05 | 4.29 |
| Iso-Valeric acid | 2 | 1.73 | 1.77 | 13.40 | 5 | 4.87 | 3.87 | 2.56 | 10 | 9.57 | 2.27 | 4.32 |
| Valeric acid | 0.5 | 0.75 | 14.92 | 50.56 | 1.25 | 1.40 | 11.03 | 11.66 | 2.5 | 2.55 | 10.45 | 1.90 |
| 4-Methylvaleric Acid | 4 | 0.34 | 5.27 | 91.56 | 10 | 7.81 | 2.85 | 21.92 | 10 | 19.51 | 3.68 | 2.43 |
| Hexanoic acid | 5 | 0.00 | 6.07 | 0.00 | 12.5 | 6.46 | 6.12 | 48.35 | 25 | 24.09 | 4.13 | 3.66 |
| Analyte | L4 | L5 | L6 | |||||||||
| C (µg/mL) | BCC (µg/mL) | RSD (%) | Accuracy RE (%) | C (µg/mL) | BCC (µg/mL) | RSD (%) | Accuracy RE (%) | C (µg/mL) | BCC (µg/mL) | RSD (%) | Accuracy RE (%) | |
| Acetic acid | 75 | 75.30 | 1.75 | 0.40 | 100 | 101.63 | 0.79 | 1.63 | 250 | 242.84 | 1.57 | 2.87 |
| Propionic acid | 37.5 | 40.09 | 4.36 | 6.92 | 50 | 55.12 | 3.60 | 10.23 | 125 | 128.61 | 2.96 | 2.89 |
| Butyric acid | 75 | 76.85 | 2.16 | 2.47 | 100 | 109.68 | 2.79 | 9.68 | 250 | 260.25 | 2.99 | 4.10 |
| Iso-Valeric acid | 15 | 14.63 | 2.19 | 2.49 | 20 | 20.26 | 0.72 | 1.28 | 50 | 50.41 | 1.47 | 0.82 |
| Valeric acid | 3.75 | 3.68 | 5.66 | 1.81 | 5 | 4.90 | 0.85 | 2.04 | 12.5 | 12.02 | 1.36 | 3.85 |
| 4-Methylvaleric Acid | 30 | 31.36 | 1.43 | 4.52 | 40 | 44.76 | 1.67 | 11.91 | 100 | 102.43 | 2.43 | 2.43 |
| Hexanoic acid | 37.5 | 42.30 | 1.76 | 12.80 | 50 | 63.43 | 1.06 | 26.86 | 125 | 130.634 | 1.43 | 4.51 |
| Analyte | L7 | L8 | ||||||||||
| C (µg/mL) | BCC (µg/mL) | RSD (%) | Accuracy RE (%) | C (µg/mL) | BCC (µg/mL) | RSD (%) | Accuracy RE (%) | |||||
| Acetic acid | 500 | 504.37 | 1.35 | 0.87 | 800 | 799.21 | 1.01 | 0.10 | ||||
| Propionic acid | 250 | 259.24 | 1.75 | 3.70 | 400 | 392.54 | 1.69 | 1.87 | ||||
| Butyric acid | 500 | 493.29 | 3.16 | 1.34 | 800 | 2.78 | ||||||
| Iso-Valeric acid | 100 | 101.67 | 2.83 | 1.67 | 160 | 158.87 | 2.00 | 0.71 | ||||
| Valeric acid | 25 | 25.12 | 1.65 | 0.49 | 40 | 40.08 | 1.59 | 0.21 | ||||
| 4-Methylvaleric Acid | 200 | 197.86 | 0.72 | 1.07 | 320 | 1.25 | ||||||
| Hexanoic acid | 250 | 244.40 | 1.37 | 2.24 | 400 | 1.84 | ||||||
| Caecum | Plasma | |||||||
|---|---|---|---|---|---|---|---|---|
| Con Calculated (µg/mL) | Con Measured (µg/mL) | Accuracy (RE%) | Precision (CV%) | Con Calculated (µg/mL) | Con Measured (µg/mL) | Accuracy (RE%) | Precision (CV%) | |
| Acetic acid | 1835.92 | 1853.92 | 0.98 | 9.93 | 107.94 | 97.55 | 9.62 | 15.64 |
| Propionic acid | 596.94 | 373.73 | 37.39 | 7.34 | 3.89 | n.d. | N/A | N/A |
| Butyric acid | 759.23 | 584.72 | 22.98 | 7.49 | 0.00 | n.d. | N/A | N/A |
| Iso-Valeric acid | 15.02 | 11.07 | 26.28 | 10.49 | 0.00 | 6.12 | 6.12 | 5.88 |
| Valeric acid | 37.41 | 28.68 | 23.33 | 7.87 | 0.21 | 0.17 | 18.45 | 8.65 |
| 4-Methylvaleric Acid | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Hexanoic acid | 1835.92 | 1853.92 | 0.98 | 9.93 | 107.94 | 97.55 | 9.62 | 15.64 |

| Analyte | Phosphoric Acid | Succinic Acid |
|---|---|---|
| Acetic acid | 177.41 | 102.9 |
| Propionic acid | 124.69 | 101.08 |
| Butyric acid | 111.82 | 95.98 |
| Iso-Valeric acid | 124.68 | 110.98 |
| Valeric acid | 121.83 | 115.26 |
| 4-Methylvaleric Acid | 114.09 | 105.8 |
| Hexanoic acid | 118.4 | 104 |
| Tissue | Analyte | Regression Linear Equation | R2 | Concentration Calculated by External Calibration (µg/mL) | Concentration Calculated by Standard Addition Method (µg/mL) |
|---|---|---|---|---|---|
| Cecum | Acetic acid | 1.5317x + 465.4086 | 0.9885 | 484.91 | 303.85 |
| Propionic acid | 0.6165x + 129.5131 | 0.9841 | 128.92 | 210.1 | |
| Butyric acid | 0.8578x + 181.2958 | 0.9896 | 184.96 | 211.35 | |
| Iso-Valeric acid | Y = 0.9646x + 3.0423 | 0.9855 | 3.51 | 3.53 | |
| Valeric acid | Y = 0.9111x + 8.3260 | 0.9857 | 8.61 | 9.14 | |
| Plasma | Acetic acid | Y = 2.4065x + 24.3615 | 0.9884 | 28.08 | 10.12 |
| Feces | Acetic acid | Y = 1.9184x + 76.8450 | 0.9694 | 87.4 | 40.06 |
| Propionic acid | Y = 1.4545 + 22.1196 | 0.9291 | 20.54 | 15.21 | |
| Butyric acid | Y = 1.1192x + 23.013 | 0.9821 | 25.48 | 20.56 | |
| Iso-Valeric acid | Y = 0.9171x + 1.4114 | 0.9724 | 1.50 | 1.54 | |
| Valeric acid | Y = 0.8817x + 3.4704 | 0.9774 | 3.68 | 3.94 | |
| Liver | Acetic acid | Y = 2.6509x + 85.0558 | 0.9840 | 92.86 | 32.09 |
| Propionic acid | Y = 1.9455x − 0.2261 | 0.8621 | 0 | 0.12 | |
| Valeric acid | Y = 1.3085x − 2.5070 | 0.9760 | 0.33 | 1.92 |
| Analyte | Plasma | Feces | Cecum | Liver | Adipose Tissue | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Low | High | Low | High | Low | High | Low | High | Low | High | |
| Acetic acid | 11.91 | 4.37 | 34.49 | 8.54 | 30.32 | 12.79 | 4.43 | 2.20 | 2.91 | 1.22 |
| Propionic acid | 2.11 | 1.63 | 9.40 | 4.96 | 9.55 | 3.47 | 1.57 | 3.11 | 1.81 | 2.99 |
| Butyric acid | 4.58 | 1.11 | 8.16 | 2.53 | 33.28 | 9.41 | 3.34 | 1.54 | 4.66 | 1.85 |
| Iso-Valeric acid | 4.84 | 7.09 | 7.09 | 6.51 | 3.71 | 5.66 | 4.99 | 6.69 | 6.06 | 4.66 |
| Valeric acid | 1.39 | 2.30 | 31.28 | 4.91 | 12.84 | 5.09 | 3.59 | 2.77 | 3.85 | 7.43 |
| 4-Methylvaleric Acid | 1.09 | 2.24 | 2.87 | 1.69 | 3.94 | 1.77 | 3.55 | 2.78 | 3.12 | 5.75 |
| Hexanoic acid | 2.76 | 3.13 | 3.04 | 3.07 | 3.17 | 2.52 | 4.59 | 3.86 | 5.05 | 6.60 |
| Analyte | Feces | Plasma |
|---|---|---|
| Acetic acid | 8.0 | 14.2 |
| Propionic acid | 4.7 | 25.3 |
| Butyric acid | 1.4 | n.d. |
| Iso-Valeric acid | 3.6 | 44.0 |
| Valeric acid | 2.0 | 49.5 |
| 4-Methylvaleric Acid | n.d. | n.d. |
| Hexanoic acid | 4.2 | n.d. |
References
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yi, C.X.; Katiraei, S.; Kooijman, S.; Zhou, E.; Chung, C.K.; Gao, Y.; van den Heuvel, J.K.; Meijer, O.C.; Berbee, J.F.P.; et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 2018, 67, 1269–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.J.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 2015, 114, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, T.; Sugitate, K.; Nakai, T.; Jikumaru, Y.; Ishihara, G. Rapid profiling method for mammalian feces short chain fatty acids by GC-MS. Anal. Biochem. 2018, 543, 51–54. [Google Scholar] [CrossRef]
- Hoving, L.R.; Heijink, M.; van Harmelen, V.; van Dijk, K.W.; Giera, M. GC-MS Analysis of Short-Chain Fatty Acids in Feces, Cecum Content, and Blood Samples. Methods Mol. Biol. 2018, 1730, 247–256. [Google Scholar] [CrossRef]
- Douny, C.; Dufourny, S.; Brose, F.; Verachtert, P.; Rondia, P.; Lebrun, S.; Marzorati, M.; Everaert, N.; Delcenserie, V.; Scippo, M.L. Development of an analytical method to detect short-chain fatty acids by SPME-GC-MS in samples coming from an in vitro gastrointestinal model. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1124, 188–196. [Google Scholar] [CrossRef]
- Rahman, M.N.; Diantini, A.; Fattah, M.; Barliana, M.I.; Wijaya, A. A highly sensitive, simple, and fast gas chromatography-mass spectrometry method for the quantification of serum short-chain fatty acids and their potential features in central obesity. Anal. Bioanal. Chem. 2021, 413, 6837–6844. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Nyman, M.; Jonsson, J.A. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed. Chromatogr. BMC 2006, 20, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Villalba, R.; Gimenez-Bastida, J.A.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A.; Carlos Espin, J.; Larrosa, M. Alternative method for gas chromatography-mass spectrometry analysis of short-chain fatty acids in faecal samples. J. Sep. Sci. 2012, 35, 1906–1913. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.; Gustavsen, S.; Nguyen, T.D.; Nyman, M.; Langkilde, A.R.; Hansen, T.H.; Sellebjerg, F.; Oturai, A.B.; Bach Sondergaard, H. Serum Short-Chain Fatty Acids and Associations With Inflammation in Newly Diagnosed Patients With Multiple Sclerosis and Healthy Controls. Front. Immunol. 2021, 12, 661493. [Google Scholar] [CrossRef] [PubMed]
- EMA. Guideline on Bioanalytical Method Validation. EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2 2009. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 17 December 2021).
- Zhang, S.; Wang, H.; Zhu, M.J. A sensitive GC/MS detection method for analyzing microbial metabolites short chain fatty acids in fecal and serum samples. Talanta 2019, 196, 249–254. [Google Scholar] [CrossRef]
- Anwer, M.S.; Stieger, B. Sodium-dependent bile salt transporters of the SLC10A transporter family: More than solute transporters. Pflug. Arch. Eur. J. Physiol. 2014, 466, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D. Microbiota and metabolites in metabolic diseases. Nat. Rev. Endocrinol. 2019, 15, 69–70. [Google Scholar] [CrossRef]
- Pauly, M.J.; Rohde, J.K.; John, C.; Evangelakos, I.; Koop, A.C.; Pertzborn, P.; Todter, K.; Scheja, L.; Heeren, J.; Worthmann, A. Inulin Supplementation Disturbs Hepatic Cholesterol and Bile Acid Metabolism Independent from Housing Temperature. Nutrients 2020, 12, 3200. [Google Scholar] [CrossRef]
- Hoverstad, T.; Midtvedt, T. Short-chain fatty acids in germfree mice and rats. J. Nutr. 1986, 116, 1772–1776. [Google Scholar] [CrossRef]
- Clausen, M.R.; Bonnen, H.; Tvede, M.; Mortensen, P.B. Colonic fermentation to short-chain fatty acids is decreased in antibiotic-associated diarrhea. Gastroenterology 1991, 101, 1497–1504. [Google Scholar] [CrossRef]
- Zarrinpar, A.; Chaix, A.; Xu, Z.Z.; Chang, M.W.; Marotz, C.A.; Saghatelian, A.; Knight, R.; Panda, S. Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat. Commun. 2018, 9, 2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnusson, B.; Örnemark, U. Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; Eurachem: Teddington, UK, 2014; Available online: https://www.eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_EN.pdf (accessed on 17 December 2021).




| Analyte | m/z (TI, CI1, CI2) | RT (min) | Regression Linear Equation | R2 | LOD (µg/mL) | LOQ (µg/mL) | Linearity Ranges (µg/mL) | Carry Over (% LOQ) |
|---|---|---|---|---|---|---|---|---|
| Acetic acid | 43, 45, 60 | 5.4 | Y = 0.0029x + 0.4198 | 0.9999 | 0.5 | 10 | 10–800 | 1.0006 |
| Propionic acid | 29, 45, 74 | 6.5 | Y = 0.0017x + 0.9075 | 0.9974 | 1 | 5 | 5–400 | 0.2535 |
| Butyric acid | 41, 60, 73 | 7.5 | Y = 0.0006x + 0.0083 | 0.9965 | 0.13 | 10 | 10–500 | 7.8136 |
| Iso-Valeric acid | 43, 60, 87 | 8.3 | Y = 0.0019x + 0.0004 | 0.9998 | 0.2 | 2 | 2–160 | 6.3859 |
| Valeric acid | 41, 60, 73 | 9.4 | Y = 0.002x − 0.0005 | 0.9998 | 0.01 | 0.5 | 0.5–40 | 5.5461 |
| 4-Methylvaleric Acid | 43, 57, 74 | 10.1 | Y = 0.0006x + 0.0030 | 0.9981 | 0.05 | 4 | 4–200 | 5.9567 |
| Hexanoic acid | 60, 73, 87 | 10.4 | Y = 0.0009x + 0.0130 | 0.9906 | 0.1 | 5 | 5–250 | 7.2714 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohde, J.K.; Fuh, M.M.; Evangelakos, I.; Pauly, M.J.; Schaltenberg, N.; Siracusa, F.; Gagliani, N.; Tödter, K.; Heeren, J.; Worthmann, A. A Gas Chromatography Mass Spectrometry-Based Method for the Quantification of Short Chain Fatty Acids. Metabolites 2022, 12, 170. https://doi.org/10.3390/metabo12020170
Rohde JK, Fuh MM, Evangelakos I, Pauly MJ, Schaltenberg N, Siracusa F, Gagliani N, Tödter K, Heeren J, Worthmann A. A Gas Chromatography Mass Spectrometry-Based Method for the Quantification of Short Chain Fatty Acids. Metabolites. 2022; 12(2):170. https://doi.org/10.3390/metabo12020170
Chicago/Turabian StyleRohde, Julia K., Marceline M. Fuh, Ioannis Evangelakos, Mira J. Pauly, Nicola Schaltenberg, Francesco Siracusa, Nicola Gagliani, Klaus Tödter, Joerg Heeren, and Anna Worthmann. 2022. "A Gas Chromatography Mass Spectrometry-Based Method for the Quantification of Short Chain Fatty Acids" Metabolites 12, no. 2: 170. https://doi.org/10.3390/metabo12020170
APA StyleRohde, J. K., Fuh, M. M., Evangelakos, I., Pauly, M. J., Schaltenberg, N., Siracusa, F., Gagliani, N., Tödter, K., Heeren, J., & Worthmann, A. (2022). A Gas Chromatography Mass Spectrometry-Based Method for the Quantification of Short Chain Fatty Acids. Metabolites, 12(2), 170. https://doi.org/10.3390/metabo12020170