
New Kids on the Block: Bile Salt Conjugates of Microbial Origin

 , ,
, ,  and
and 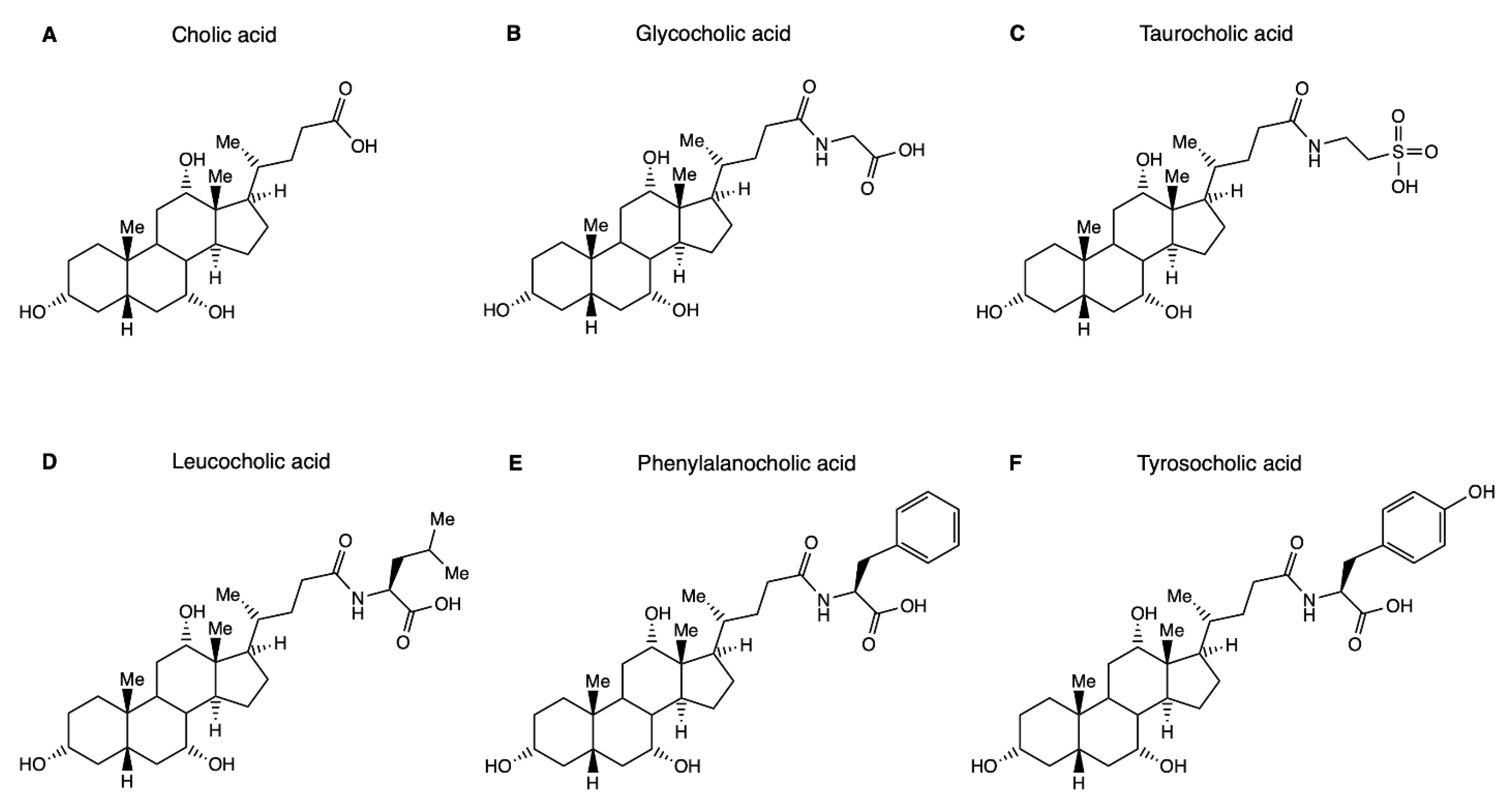{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Hepatic Bile Salt Conjugation
1.2. Microbial Bile Salt Conjugation
2. Possible Functions of MBSCs
2.1. MBSCs Are Likely Substrates for Bile Salt Transporters
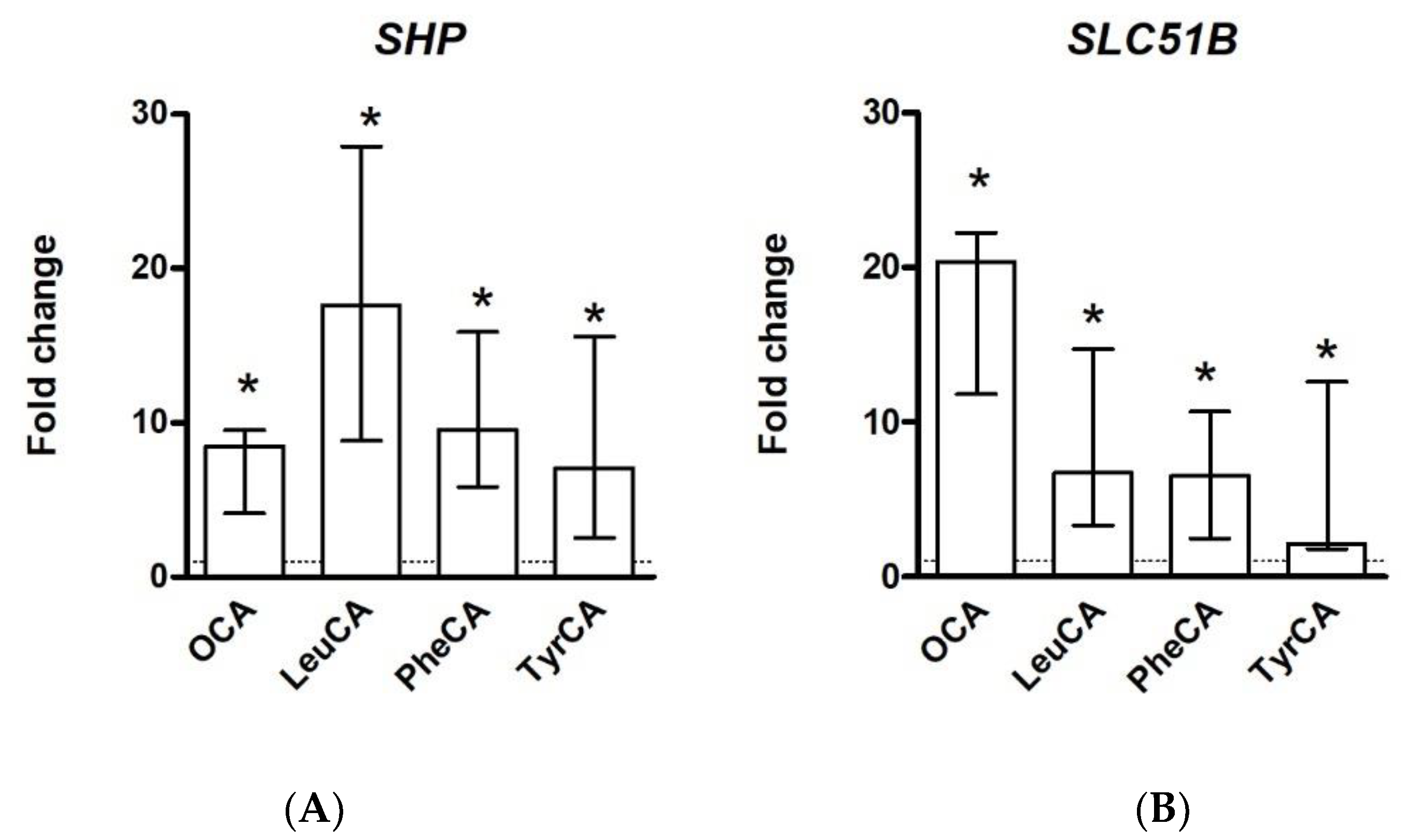
2.2. MBSCs Are Likely Ligands for Bile Salt Receptors
2.2.1. Plasma Membrane Bile Salt Receptors
2.2.2. Nuclear Bile Salt Receptors
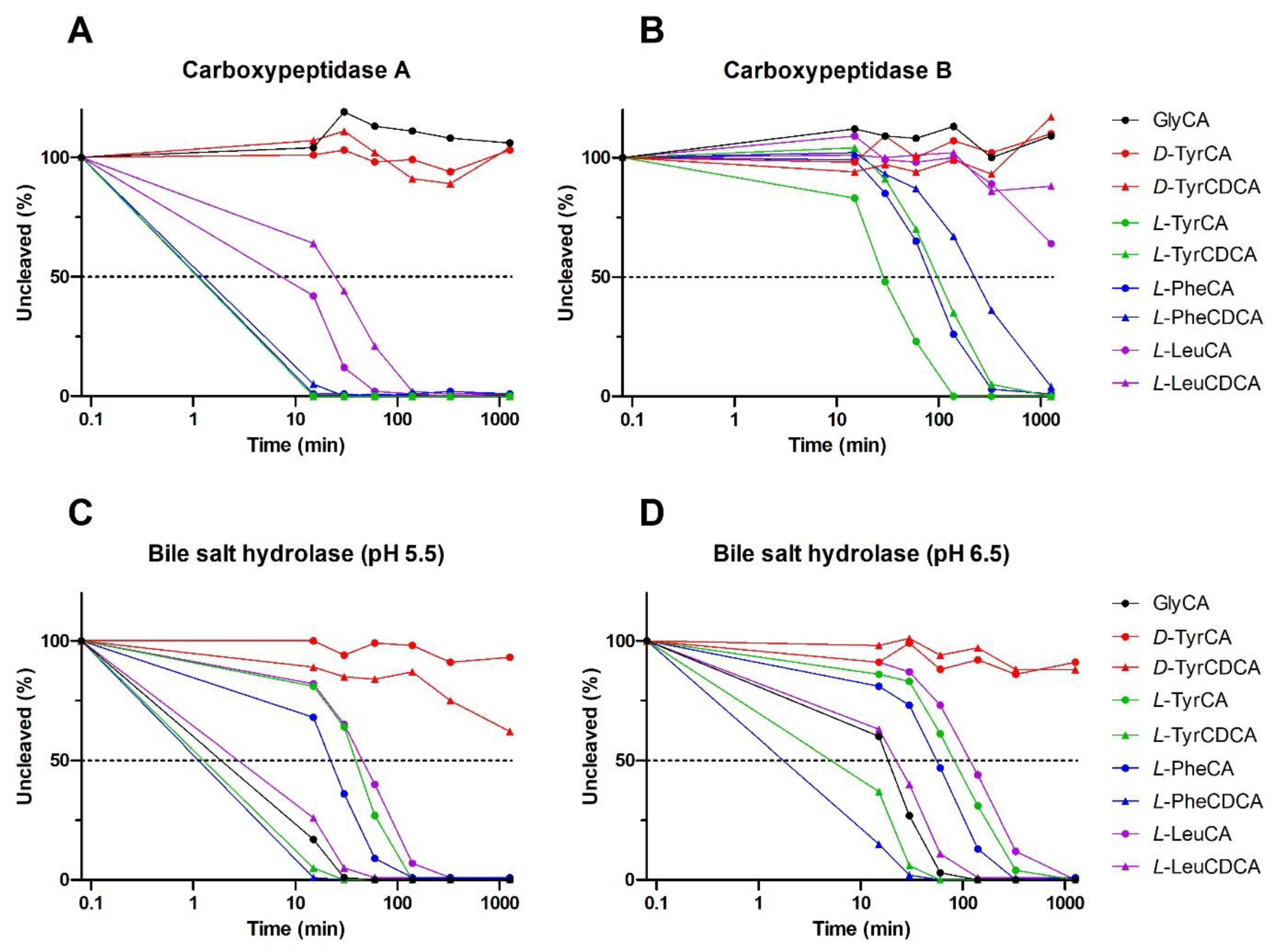
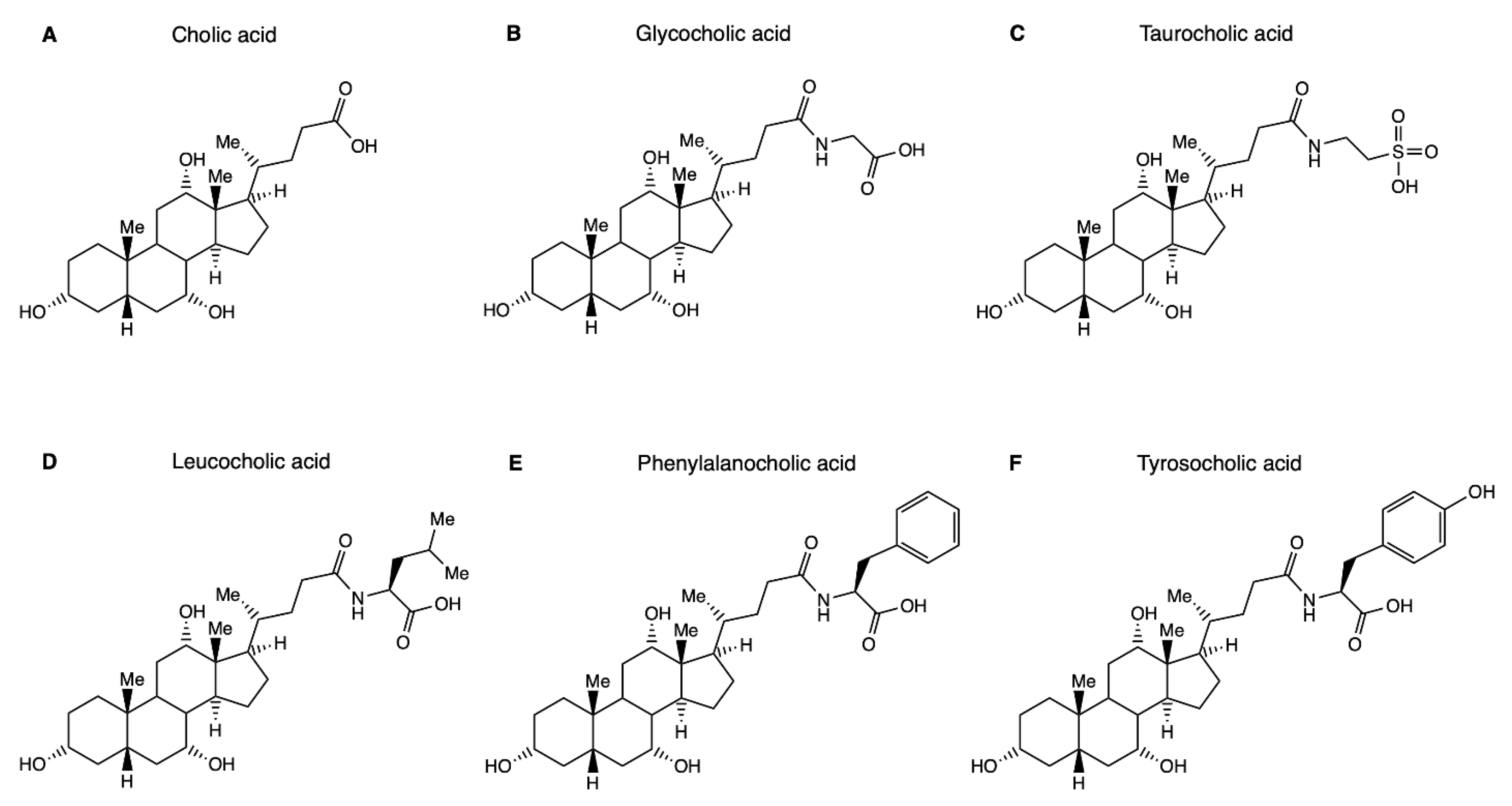
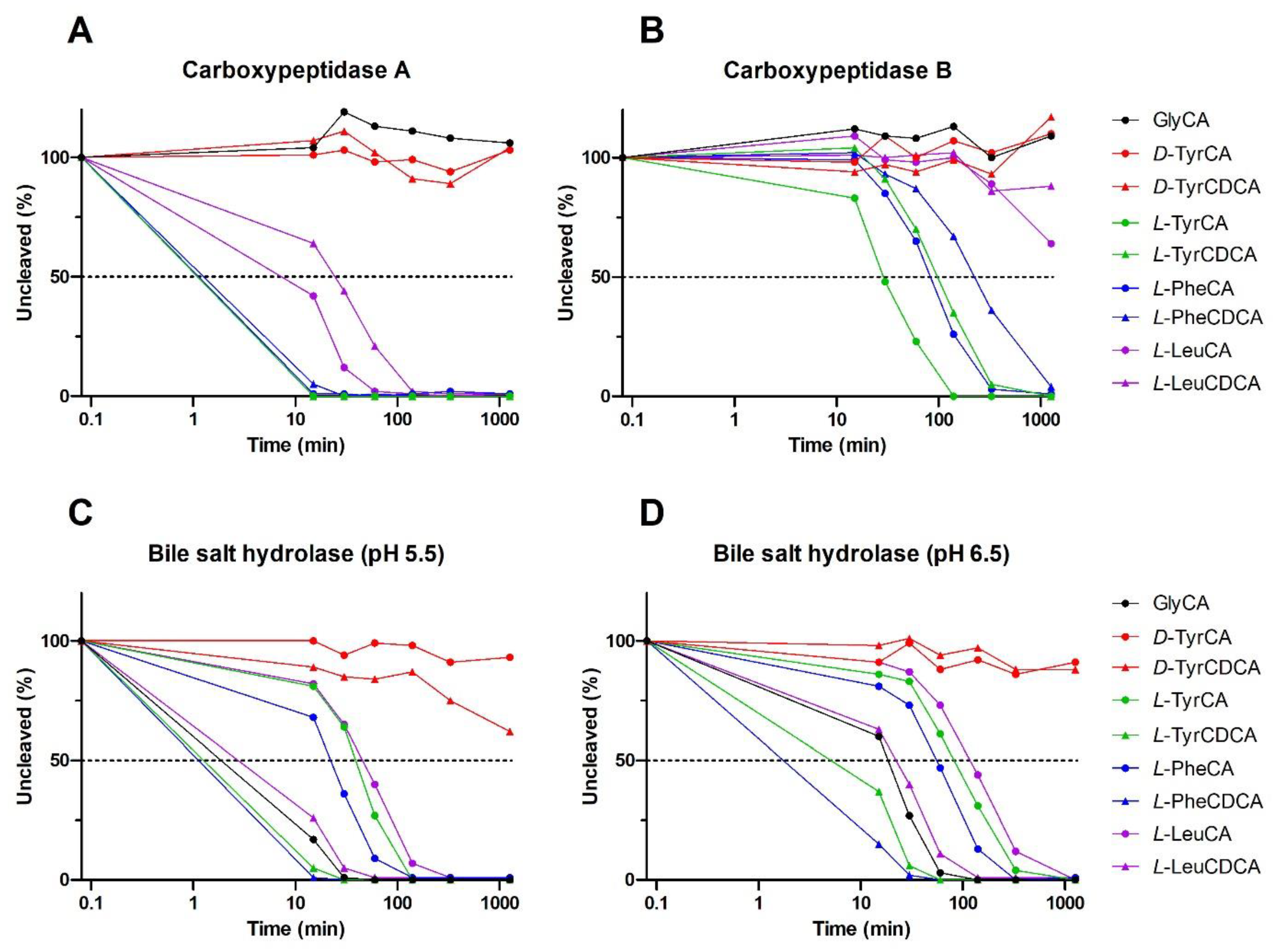
2.3. MBSCs Are Prone to Degradation by Host and Bacterial Enzymes
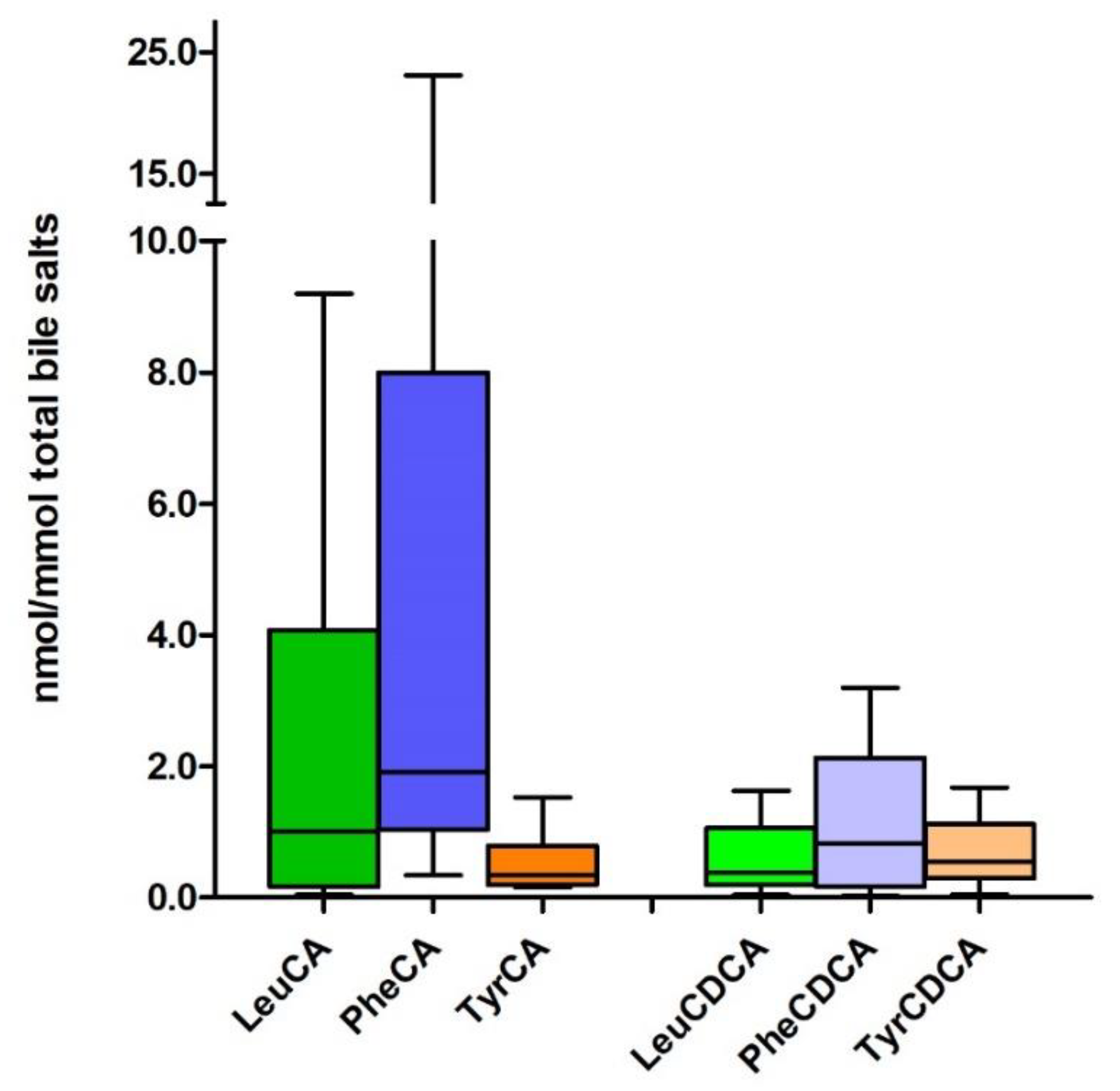
2.4. MBSCs in Human Disease and Experimental Models
2.5. Potential Roles of MBSCs in the Gut
3. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Hofmann, A.F. Biliary secretion and excretion in health and disease: Current concepts. Ann. Hepatol. 2007, 6, 15–27. [Google Scholar] [CrossRef]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philipp, B. Bacterial degradation of bile salts. Appl. Microbiol. Biotechnol. 2011, 89, 903–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.F.; Hagey, L.R.; Krasowski, M.D. Bile salts of vertebrates: Structural variation and possible evolutionary significance. J. Lipid Res. 2010, 51, 226–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huijghebaert, S.M.; Hofmann, A.F. Pancreatic carboxypeptidase hydrolysis of bile acid-amino conjugates: Selective resistance of glycine and taurine amidates. Gastroenterology 1986, 90, 306–315. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.F.; Roda, A. Physicochemical properties of bile acids and their relationship to biological properties: An overview of the problem. J. Lipid Res. 1984, 25, 1477–1489. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Quinn, R.A.; Melnik, A.V.; Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 2020, 579, 123–129. [Google Scholar] [CrossRef]
- Lucas, L.N.; Barrett, K.; Kerby, R.L.; Zhang, Q.; Cattaneo, L.E.; Stevenson, D.; Rey, F.E.; Amador-Noguez, D. Dominant Bacterial Phyla from the Human Gut Show Widespread Ability to Transform and Conjugate Bile Acids. mSystems 2021, 6, e0080521. [Google Scholar] [CrossRef]
- Mallonee, D.H.; Hylemon, P.B. Sequencing and expression of a gene encoding a bile acid transporter from Eubacterium sp. strain VPI 12708. J. Bacteriol. 1996, 178, 7053–7058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, P.A. Bile Formation and the Enterohepatic Circulation. In Physiology of the Gastrointestinal Tract, 6th ed.; Said, H.M., Ed.; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Sun, A.Q.; Balasubramaniyan, N.; Chen, H.; Shahid, M.; Suchy, F.J. Identification of functionally relevant residues of the rat ileal apical sodium-dependent bile acid cotransporter. J. Biol. Chem. 2006, 281, 16410–16418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Ma, L.; Dawson, P.A.; Sinal, C.J.; Sehayek, E.; Gonzalez, F.J.; Breslow, J.; Ananthanarayanan, M.; Shneider, B.L. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J. Biol. Chem. 2003, 278, 19909–19916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lack, L.; Weiner, I.M. Intestinal bile salt transport: Structure-activity relationships and other properties. Am. J. Physiol. 1966, 210, 1142–1152. [Google Scholar] [CrossRef] [Green Version]
- Chopyk, D.M.; Grakoui, A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology 2020, 159, 849–863. [Google Scholar] [CrossRef]
- Dawson, P.A.; Hubbert, M.L.; Rao, A. Getting the mOST from OST: Role of organic solute transporter, OSTalpha-OSTbeta, in bile acid and steroid metabolism. Biochim. Biophys. Acta 2010, 1801, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Anwer, M.S.; O’Maille, E.R.; Hofmann, A.F.; DiPietro, R.A.; Michelotti, E. Influence of side-chain charge on hepatic transport of bile acids and bile acid analogues. Am. J. Physiol. 1985, 249, G479–G488. [Google Scholar] [CrossRef]
- Stieger, B.; Meier, Y.; Meier, P.J. The bile salt export pump. Pflügers Arch.-Eur. J. Physiol. 2007, 453, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Heuman, D.M.; Pandak, W.M.; Hylemon, P.B.; Vlahcevic, Z.R. Conjugates of ursodeoxycholate protect against cytotoxicity of more hydrophobic bile salts: In vitro studies in rat hepatocytes and human erythrocytes. Hepatology 1991, 14, 920–926. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Reich, M.; Klindt, C.; Deutschmann, K.; Spomer, L.; Häussinger, D.; Keitel, V. Role of the G Protein-Coupled Bile Acid Receptor TGR5 in Liver Damage. Dig. Dis. 2017, 35, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.N.; Harris, D.A.; Aliakbarian, H.; Luo, J.N.; Henke, M.T.; Subramaniam, R.; Vernon, A.H.; Tavakkoli, A.; Sheu, E.G.; Devlin, A.S. Bariatric surgery reveals a gut-restricted TGR5 agonist with anti-diabetic effects. Nat. Chem. Biol. 2021, 17, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Macchiarulo, A.; Thomas, C.; Gioiello, A.; Une, M.; Hofmann, A.F.; Saladin, R.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. Novel potent and selective bile acid derivatives as TGR5 agonists: Biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 2008, 51, 1831–1841. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, X.; Meng, Z.; Dong, B.; Shiah, S.; Moore, D.D.; Huang, W. Significance and Mechanism of CYP7a1 Gene Regulation during the Acute Phase of Liver Regeneration. Mol. Endocrinol. 2009, 23, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Raufman, J.-P.; Cheng, K.; Zimniak, P. REVIEW: Activation of Muscarinic Receptor Signaling by Bile Acids: Physiological and Medical Implications. Dig. Dis. Sci. 2003, 48, 1431–1444. [Google Scholar] [CrossRef]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P.N. Bile acid: A potential inducer of colon cancer stem cells. Stem Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Yuza, K.; Hirose, Y.; Nakajima, M.; Ramanathan, R.; Hait, N.C.; Hylemon, P.B.; Zhou, H.; Takabe, K.; Wakai, T. The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J. Lipid Res. 2016, 57, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Keitel, V.; Stindt, J.; Häussinger, D. Bile Acid-Activated Receptors: GPBAR1 (TGR5) and Other G Protein-Coupled Receptors. Handb. Exp. Pharmacol. 2019, 256, 19–49. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Panzitt, K.; Wagner, M. FXR in liver physiology: Multiple faces to regulate liver metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166133. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Song, C.; Hiipakka, R.A.; Liao, S. Selective activation of liver X receptor alpha by 6alpha-hydroxy bile acids and analogs. Steroids 2000, 65, 423–427. [Google Scholar] [CrossRef]
- De Marino, S.; Carino, A.; Masullo, D.; Finamore, C.; Marchiano, S.; Cipriani, S.; Di Leva, F.S.; Catalanotti, B.; Novellino, E.; Limongelli, V.; et al. Hyodeoxycholic acid derivatives as liver X receptor alpha and G-protein-coupled bile acid receptor agonists. Sci. Rep. 2017, 7, 43290. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Cai, J.; Gonzalez, F.J. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 335–347. [Google Scholar] [CrossRef]
- Hollman, D.A.; Milona, A.; van Erpecum, K.J.; van Mil, S.W. Anti-inflammatory and metabolic actions of FXR: Insights into molecular mechanisms. Biochim. Biophys. Acta 2012, 1821, 1443–1452. [Google Scholar] [CrossRef]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Koelfat, K.V.K.; Picot, D.; Chang, X.; Desille-Dugast, M.; van Eijk, H.M.; van Kuijk, S.M.J.; Lenicek, M.; Layec, S.; Carsin, M.; Dussaulx, L.; et al. Chyme Reinfusion Restores the Regulatory Bile Salt-FGF19 Axis in Patients with Intestinal Failure. Hepatology 2021, 74, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Florén, C.H.; Nilsson, A. Binding of bile salts to fibre-enriched wheat bran. Hum. Nutr Clin. Nutr. 1982, 36, 381–390. [Google Scholar] [PubMed]
- Begley, M.; Gahan, C.G.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [Green Version]
- Barrasa, J.I.; Olmo, N.; Lizarbe, M.A.; Turnay, J. Bile acids in the colon, from healthy to cytotoxic molecules. Toxicol. Vitr. 2013, 27, 964–977. [Google Scholar] [CrossRef]
- Batta, A.K.; Salen, G.; Arora, R.; Shefer, S.; Batta, M.; Person, A. Side chain conjugation prevents bacterial 7-dehydroxylation of bile acids. J. Biol. Chem. 1990, 265, 10925–10928. [Google Scholar] [CrossRef]
- Marion, S.; Desharnais, L.; Studer, N.; Dong, Y.; Notter, M.D.; Poudel, S.; Menin, L.; Janowczyk, A.; Hettich, R.L.; Hapfelmeier, S.; et al. Biogeography of microbial bile acid transformations along the murine gut. J. Lipid Res. 2020, 61, 1450–1463. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 2021, 599, 458–464. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ay, Ü.; Leníček, M.; Classen, A.; Olde Damink, S.W.M.; Bolm, C.; Schaap, F.G. New Kids on the Block: Bile Salt Conjugates of Microbial Origin. Metabolites 2022, 12, 176. https://doi.org/10.3390/metabo12020176
Ay Ü, Leníček M, Classen A, Olde Damink SWM, Bolm C, Schaap FG. New Kids on the Block: Bile Salt Conjugates of Microbial Origin. Metabolites. 2022; 12(2):176. https://doi.org/10.3390/metabo12020176
Chicago/Turabian StyleAy, Ümran, Martin Leníček, Arno Classen, Steven W. M. Olde Damink, Carsten Bolm, and Frank G. Schaap. 2022. "New Kids on the Block: Bile Salt Conjugates of Microbial Origin" Metabolites 12, no. 2: 176. https://doi.org/10.3390/metabo12020176
APA StyleAy, Ü., Leníček, M., Classen, A., Olde Damink, S. W. M., Bolm, C., & Schaap, F. G. (2022). New Kids on the Block: Bile Salt Conjugates of Microbial Origin. Metabolites, 12(2), 176. https://doi.org/10.3390/metabo12020176