
Human Brain Lipidomics: Pilot Analysis of the Basal Ganglia Sphingolipidome in Parkinson’s Disease and Lewy Body Disease

Abstract
:1. Introduction
2. Results
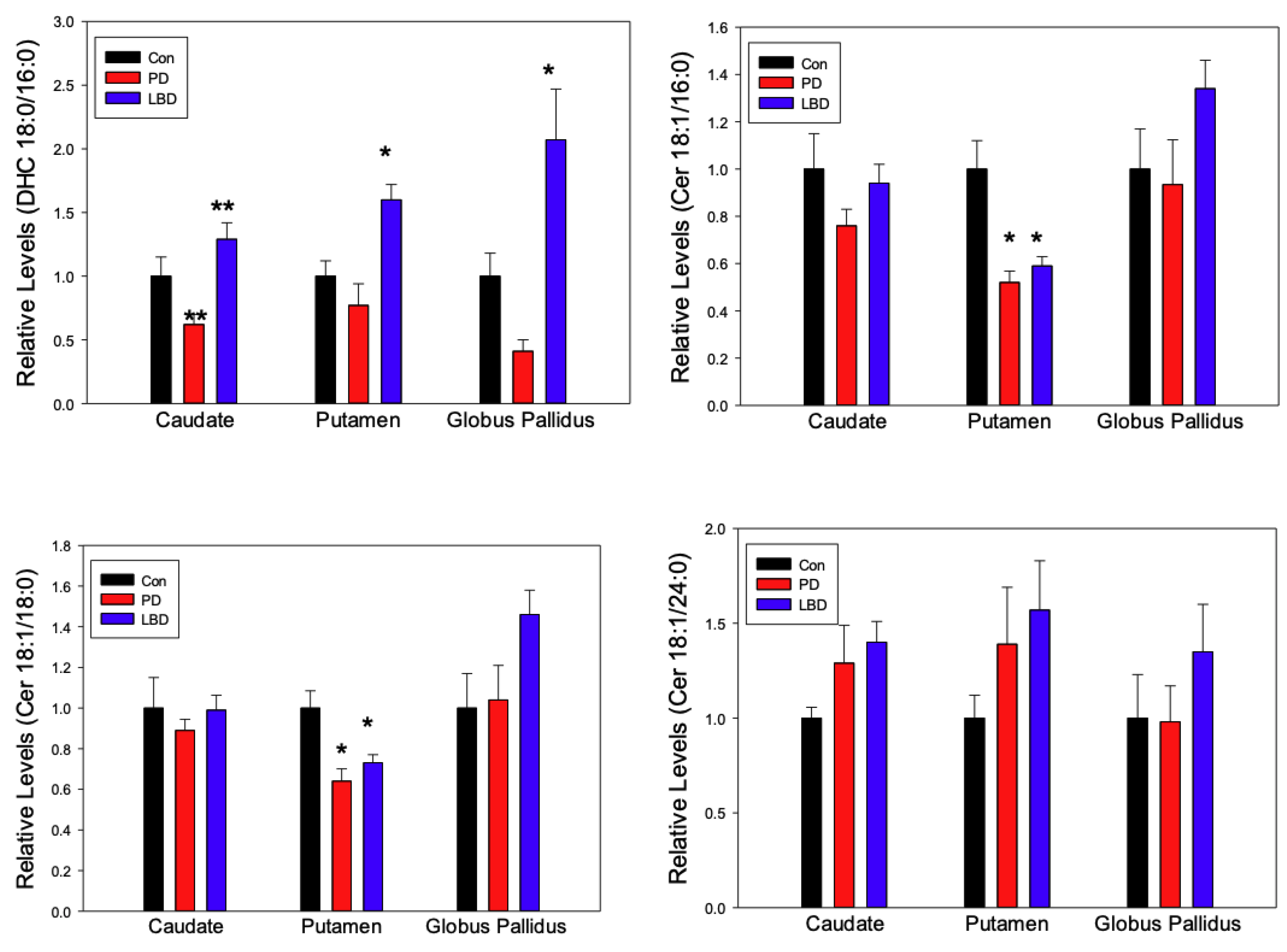
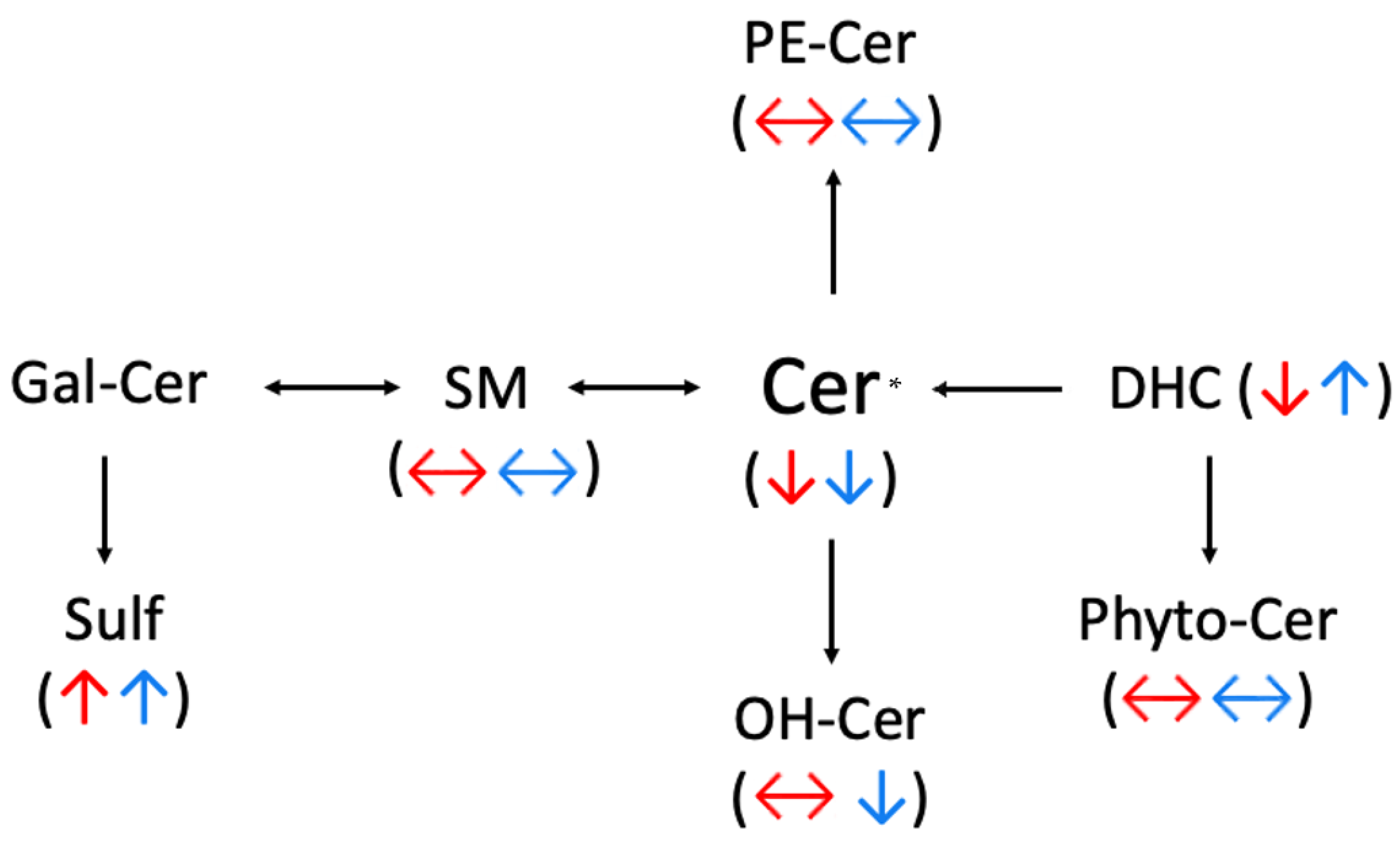
2.1. Ceramides
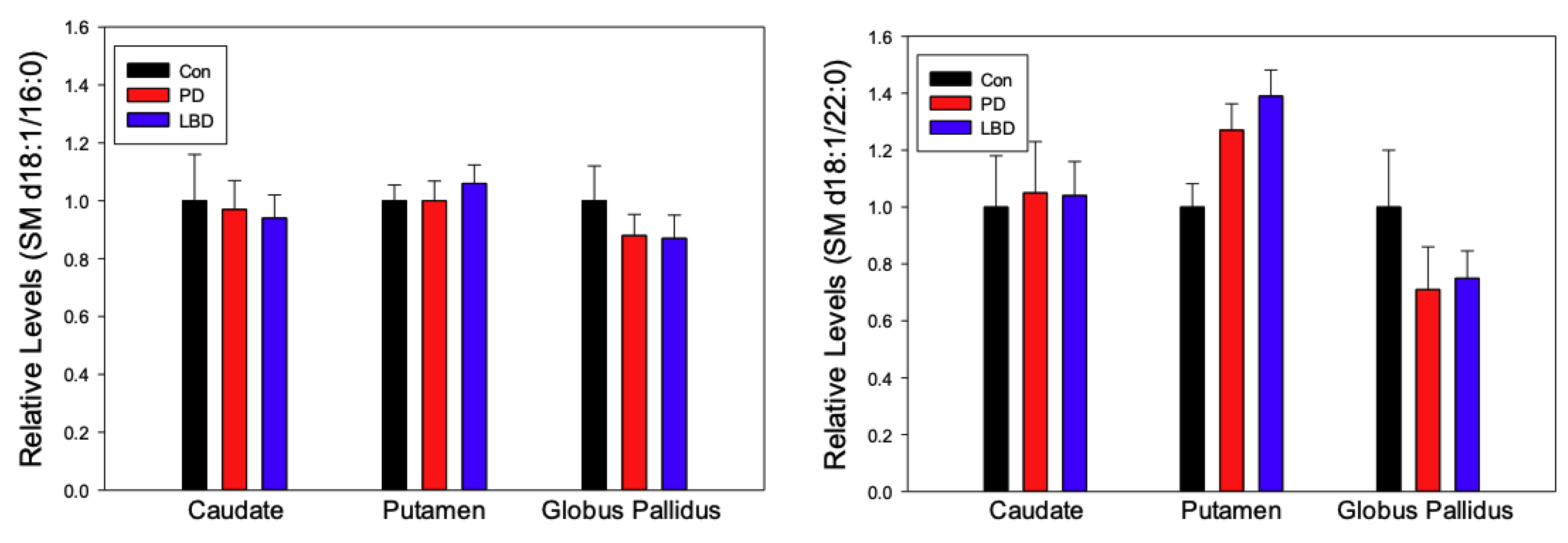
2.2. Sphingomyelins (SM)
2.3. Sulfatide (Sulf)
2.4. Gender, Age and PMI
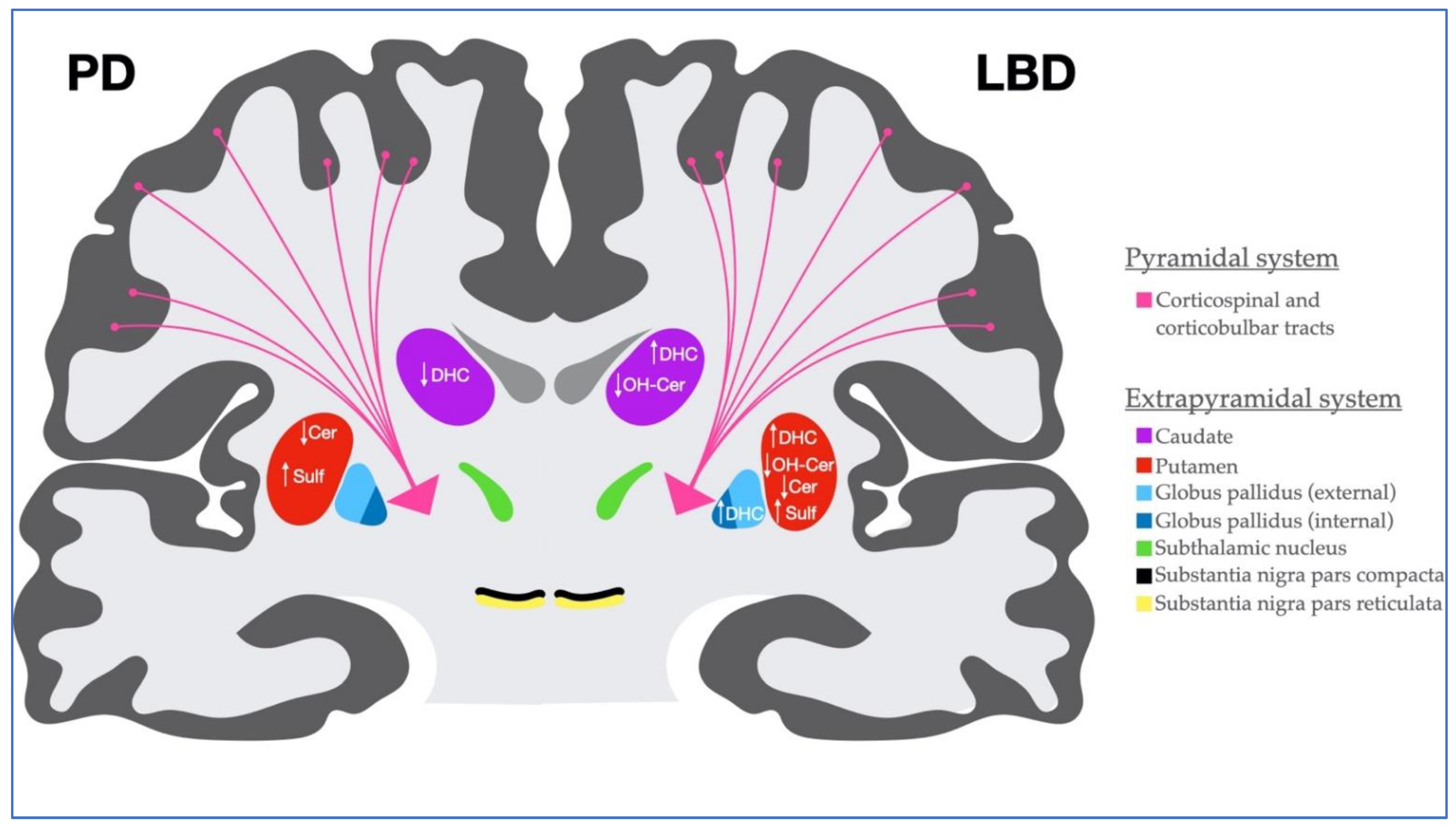
3. Discussion
4. Materials and Methods
4.1. Ethical Approval
4.2. Human Brain Samples
4.3. Lipid Extraction and Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayes, M.T. Parkinson’s disease and parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Hely, M.A.; Reid, W.G.J.; Adena, M.A.; Halliday, G.M.; Morris, J.G.L. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef]
- Kosaka, K. Lewy body disease and dementia with Lewy bodies. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Vann Jones, S.A.; O’Brien, J.T. The prevalence and incidence of dementia with Lewy bodies: A systematic review of population and clinical studies. Psychol. Med. 2014, 44, 673–683. [Google Scholar] [CrossRef]
- Sezgin, M.; Bilgic, B.; Tinaz, S.; Emre, M. Parkinson’s disease dementia and Lewy body disease. Semin. Neurol. 2019, 39, 274–282. [Google Scholar] [CrossRef]
- Henderson, M.X.; Trojanowski, J.Q.; Lee, V.M.-Y. α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Physiol. Behav. 2019, 709, 134316. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The role of lipids in Parkinson’s disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Wood, P.L.; Tippireddy, S.; Feriante, J.; Woltjer, R.L. Augmented frontal cortex diacylglycerol levels in Parkinson’s disease and Lewy body disease. PLoS ONE 2018, 13, e0191815. [Google Scholar] [CrossRef]
- Colombaioni, L.; Garcia-Gil, M. Sphingolipid metabolites in neural signalling and function. Brain Res. Rev. 2004, 46, 328–355. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Jin, H.K.; Bae, J.S. Sphingolipids in neuroinflammation: A potential target for diagnosis and therapy. BMB Rep. 2020, 53, 28–34. [Google Scholar] [CrossRef] [PubMed]
- De Wit, N.M.; Den Hoedt, S.; Martinez-Martinez, P.; Rozemuller, A.J.; Mulder, M.T.; De Vries, H.E. Astrocytic ceramide as possible indicator of neuroinflammation. J. Neuroinflamm. 2019, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.-K.; Gräber-Sultan, S.; Schleicher, E.; et al. Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: A pilot study. PLoS ONE 2013, 8, e73094. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Irigoyen, J.; Cartas-Cejudo, P.; Iruarrizaga-Lejarreta, M.; Santamaría, E. Alteration in the cerebrospinal fluid lipidome in Parkinson’s disease: A post-mortem pilot study. Biomedicines 2021, 9, 491. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Murray, M.; Persson, X.; Kantarci, K.; Parisi, J.E.; Dickson, D.W.; Petersen, R.C.; Ferman, T.J.; Boeve, B.F.; Mielke, M.M. Plasma sphingolipid changes with autopsy-confirmed Lewy body or Alzheimer’s pathology. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 3, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teitsdottir, U.D.; Halldorsson, S.; Rolfsson, O.; Lund, S.H.; Jonsdottir, M.K.; Snaedal, J.; Petersen, P.H. Cerebrospinal fluid C18 ceramide associates with markers of Alzheimer’s disease and inflammation at the pre- and early stages of dementia. J. Alzheimer’s Dis. 2021, 81, 231–244. [Google Scholar] [CrossRef]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.I.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of α-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.; Schapira, A.H.V. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Kurzawa-Akanbi, M.; Hanson, P.S.; Blain, P.G.; Lett, D.J.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M. Glucocerebrosidase mutations alter the endoplasmic reticulum and lysosomes in Lewy body disease. J. Neurochem. 2012, 123, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Henderson, M.X.; Sedor, S.; McGeary, I.; Cornblath, E.J.; Peng, C.; Riddle, D.M.; Li, H.L.; Zhang, B.; Brown, H.; Olufemi, M.F.; et al. Glucocerebrosidase activity modulates neuronal susceptibility to pathological α-synuclein insult. Neuron 2020, 5, 822–836. [Google Scholar] [CrossRef] [PubMed]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-related abnormalities in substantia nigra lipids in Parkinson’s disease. ASN Neuro 2018, 10, 1759091418781889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzawa-Akanbi, M.; Tammireddy, S.; Fabrik, I.; Gliaudelytė, L.; Doherty, M.K.; Heap, R.; Matečko-Burmann, I.; Burmann, B.M.; Trost, M.; Lucocq, J.M.; et al. Altered ceramide metabolism is a feature in the extracellular vesicle-mediated spread of alpha-synuclein in Lewy body disorders. Acta Neuropathol. 2021, 142, 961–984. [Google Scholar] [CrossRef]
- Bostan, A.C.; Dum, R.P.; Strick, P.L. Functional anatomy of basal ganglia circuits with the cerebral cortex and the cerebellum. Prog. Neurol. Surg. 2018, 33, 50–61. [Google Scholar]
- Vitanova, K.S.; Stringer, K.M.; Benitez, D.P.; Brenton, J.; Cummings, D.M. Dementia associated with disorders of the basal ganglia. J. Neurosci. Res. 2019, 97, 1728–1741. [Google Scholar] [CrossRef]
- Draganski, B.; Kherif, F.; Klöppel, S.; Cook, P.A.; Alexander, D.C.; Parker, G.; Deichmann, R.; Ashburner, J.; Frackowiak, R. Evidence for segregated and integrative connectivity patterns in the human basal ganglia. J. Neurosci. 2008, 28, 7143–7152. [Google Scholar] [CrossRef] [Green Version]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- Day, M.; Wang, Z.; Ding, J.; An, X.; Ingham, C.A.; Shering, A.F.; Wokosin, D.; Ilijic, E.; Sun, Z.; Sampson, A.R.; et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 2006, 9, 251–259. [Google Scholar] [CrossRef]
- Pagonabarraga, J.; Kulisevsky, J. Cognitive impairment and dementia in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Hauther, K.A.; Scarborough, J.H.; Craney, D.J.; Dudzik, B.; Cebak, J.E.; Woltjer, R.L. Human brain lipidomics: Utilities of chloride adducts in flow injection analysis (FIA). Life 2021, 11, 403. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Jenner, A.M.; Shui, G.; Cheong, W.F.; Mitchell, T.W.; Nealon, J.R.; Kim, W.S.; McCann, H.; Wenk, M.R.; Halliday, G.M. Lipid pathway alterations in Parkinson’s disease primary visual cortex. PLoS ONE 2011, 6, e17299. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.C.; Lee, Y.; Moon, S.; Ryu, J.H.; Oh, S. Phytoceramide shows neuroprotection and ameliorates scopolamine-induced memory impairment. Molecules 2011, 16, 9090–9100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Jang, J.Y.; Yoo, H.S.; Seong, Y.H. Neuroprotective effect of phytoceramide against transient focal ischemia-induced brain damage in rats. Arch. Pharm. Res. 2015, 38, 2241–2250. [Google Scholar] [CrossRef]
- Bickert, A.; Ginkel, C.; Kol, M.; Vom Dorp, K.; Jastrow, H.; Degen, J.; Jacobs, R.L.; Vance, D.E.; Winterhager, E.; Jiang, X.-C.; et al. Functional characterization of enzymes catalyzing ceramide phosphoethanolamine biosynthesis in mice. J. Lipid Res. 2015, 56, 821–835. [Google Scholar] [CrossRef] [Green Version]
- Ternes, P.; Brouwers, J.F.H.M.; van den Dikkenberg, J.; Holthuis, J.C.M. Sphingomyelin synthase SMS2 displays dual activity as ceramide phosphoethanolamine synthase. J. Lipid Res. 2009, 50, 2270–2277. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H.V. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 2015, 30, 1085–1089. [Google Scholar] [CrossRef]
- Lachkar, F.; Ferre, P.; Foufelle, F.; Papaioannou, A. Dihydroceramides: Their emerging physiological roles and functions in cancer and metabolic diseases. Am. J. Physiol. Endocrinol. Metab. 2021, 320, 122–130. [Google Scholar] [CrossRef]
- Ordóñez-Gutiérrez, L.; Benito-Cuesta, I.; Abad, J.L.; Casas, J.; Fábrias, G.; Wandosell, F. Dihydroceramide desaturase 1 inhibitors reduce amyloid-β levels in primary neurons from an Alzheimer’s disease transgenic model. Pharm. Res. 2018, 35, 49. [Google Scholar] [CrossRef]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Stafanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2675154/pdf/cam0301_0088.pdf (accessed on 13 January 2009). [CrossRef] [PubMed] [Green Version]
- Idkowiak-Baldys, J.; Apraiz, A.; Li, L.; Rahmaniyan, M.; Clarke, C.J.; Kraveka, J.M.; Asumendi, A.; Hannun, Y.A. Dihydroceramide desaturase activity is modulated by oxidative stress. Biochem. J. 2010, 427, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzam, R.; Hariri, F.; El-Hachem, N.; Kamar, A.; Dbaibo, G.; Nemer, G.; Bitar, F. Regulation of de novo ceramide synthesis: The role of dihydroceramide desaturase and transcriptional factors NFATC and Hand2 in the hypoxic mouse heart. DNA Cell Biol. 2013, 32, 310–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraveka, J.M.; Li, L.; Szulc, Z.M.; Bielawski, J.; Ogretmen, B.; Hannun, Y.A.; Obeid, L.; Bielawska, A. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J. Biol. Chem. 2007, 282, 16718–16728. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, P.; Knight, W.; Guo, Y.; Perlmutter, J.S.; Benzinger, T.L.S.; Morris, J.C.; Xu, J. The interactions of dopamine and oxidative damage in the striatum of patients with neurodegenerative diseases. J. Neurochem. 2020, 152, 235–251. [Google Scholar] [CrossRef] [Green Version]
- Mencarelli, C.; Martinez-Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef] [Green Version]
- Bras, J.; Singleton, A.; Cookson, M.R.; Hardy, J. Emerging pathways in genetic Parkinson’s disease: Potential role of ceramide metabolism in Lewy body disease. FEBS J. 2008, 275, 5767–5773. [Google Scholar] [CrossRef]
- Haughey, N.J. Sphingolipids in neurodegeneration. NeuroMol. Med. 2010, 12, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Grösch, S.; Schiffmann, S.; Geisslinger, G. Chain length-specific properties of ceramides. Prog. Lipid Res. 2012, 51, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson’s disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Custodia, A.; Aramburu-Núñez, M.; Correa-Paz, C.; Posado-Fernández, A.; Gómez-Larrauri, A.; Castillo, J.; Gómez-Muñoz, A.; Sobrino, T.; Ouro, A. Ceramide metabolism and Parkinson’s disease—Therapeutic targets. Biomolecules 2021, 11, 945. [Google Scholar] [CrossRef]
- Ryan, E.; Seehra, G.; Sharma, P.; Sidransky, E. GBA1-associated parkinsonism: New insights and therapeutic opportunities. Curr. Opin. Neurol. 2019, 32, 589–596. [Google Scholar] [CrossRef]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.-H.; Sun, Y.; Tomlinson, J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid β-glucosidase mutants linked to gaucher disease, parkinson disease, and lewy body dementia alter α-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, A.; Saddoughi, S.A.; Song, P.; Sultan, I.; Ponnusamy, S.; Senkal, C.E.; Snook, C.F.; Arnold, H.K.; Sears, R.C.; Hanniui, Y.A.; et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009, 23, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Du, T.-T.; Wang, L.; Duan, C.-L.; Lu, L.-L.; Zhang, J.-L.; Gao, G.; Qiu, X.-B.; Wang, X.-M.; Yang, H. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.F.; Kachergus, J.M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Lynch, T.; Hulihan, M.M.; Cobb, S.A.; Wu, R.-M.; Lu, C.-S.; et al. Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics 2005, 6, 171–177. [Google Scholar] [CrossRef]
- Zabetian, C.P.; Samii, A.; Mosley, A.D.; Roberts, J.W.; Leis, B.C.; Yearout, D.; Raskind, W.H.; Griffith, A. A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 2005, 65, 741–744. [Google Scholar] [CrossRef]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinkle, K.M.; Yue, M.; Behrouz, B.; Dächsel, J.C.; Lincoln, S.J.; Bowles, E.E.; Beevers, J.E.; Dugger, B.; Winner, B.; Prots, I.; et al. LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor co-ordination behaviors. Mol. Neurodegener. 2012, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyogashima, M.; Tadano-Aritomi, K.; Aoyama, T.; Yusa, A.; Goto, Y.; Tamiya-Koizumi, K.; Ito, H.; Murate, T.; Kannagi, R.; Hara, A. Chemical and apoptotic properties of hydroxy-ceramides containing long-chain bases with unusual alkyl chain lengths. J. Biochem. 2008, 144, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Hama, H. Fatty acid 2-Hydroxylation in mammalian sphingolipid biology. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Foo, J.-N.; Liany, H.; Bei, J.-X.; Yu, X.-Q.; Liu, J.; Au, W.-L.; Prakash, K.M.; Tan, L.C.; Tan, E.-K. A rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol. Aging 2013, 34, 2890.e13–2890.e15. [Google Scholar] [CrossRef]
- Den Hartog Jager, W.A. Sphingomyelin in lewy inclusion bodies in Parkinson’s disease. Arch. Neurol. 1969, 21, 615–619. [Google Scholar] [CrossRef]
- Jeon, S.-B.; Yoon, H.J.; Park, S.-H.; Kim, I.-H.; Park, E.J. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J. Immunol. 2008, 181, 8077–8087. [Google Scholar] [CrossRef]
- Martinelli, P.; Ippoliti, M.; Montanari, M.; Martinelli, A.; Mochi, M.; Giuliani, S. Arylsulphatase A (ASA) activity in parkinsonism and symptomatic essential tremor. Acta Neurol. Scand. 1994, 89, 171–174. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Wood, P.L. Non-targeted lipidomics utilizing constant infusion high resolution ESI mass spectrometry. In Lipidomics; Humana Press: New York, NY, USA, 2017; pp. 13–19. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Controls | PD | LBD |
|---|---|---|---|
| n | 9 | 7 | 14 |
| Age (Yrs † ± SD) [Range] | 68.25 ± 11.06 [52–81] | 88.83 ± 7.11 [78–100] | 81.42 ± 9.47 [64–93] |
| PMI (Hrs */Yrs † ± SD) [Range] | 17.75 ± 4.46 [11–23] | 18.88 ± 19.1 [2–52] | 10.13 ± 8.58 [3–32] |
| Sex (M:F) | 5:3 | 4:3 | 9:5 |
| Neuritic plaques (sparse) n | 2 | 3 | 11 |
| Neuritic plaques (moderate) n | 0 | 1 | 3 |
| Tangles Braak 1 n | 5 | 3 | 2 |
| Tangles Braak 2 n | 0 | 1 | 5 |
| Tangles Braak 3 n | 0 | 0 | 1 |
| Tangles Braak 4 n | 0 | 3 | 6 |
| Cortical Lewy Bodies n | 0 | 7 | 14 |
| Lipid | Exact Mass | [M + Cl]− |
|---|---|---|
| DHC d18:0/16:0 | 539.5272 | 574.4975 |
| Cer d18:1/16:0 | 537.5121 | 572.4819 |
| Cer d18:1/18:0 | 656.5434 | 600.5132 |
| Cer d18:1/24:0 | 649.6373 | 684.6071 |
| OH-Cer d18:1/18:0 | 581.5382 | 616.5082 |
| OH-Cer d18:1/24:0 | 665.6322 | 700.6021 |
| PhytoCer t18:0/18:0 | 583.5540 | 618.5238 |
| PE-Cer d18:1/18:0 | 772.6458 | 807.6156 |
| SM d18:1/16:0 | 704.5832 | 739.5531 |
| SM d18:1/22:0 | 788.6771 | 823.6470 |
| Lipid | Exact Mass | [M−H]− |
| Sulf d18:1/24:0 | 863.6156 | 862.6082 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beger, A.W.; Dudzik, B.; Woltjer, R.L.; Wood, P.L. Human Brain Lipidomics: Pilot Analysis of the Basal Ganglia Sphingolipidome in Parkinson’s Disease and Lewy Body Disease. Metabolites 2022, 12, 187. https://doi.org/10.3390/metabo12020187
Beger AW, Dudzik B, Woltjer RL, Wood PL. Human Brain Lipidomics: Pilot Analysis of the Basal Ganglia Sphingolipidome in Parkinson’s Disease and Lewy Body Disease. Metabolites. 2022; 12(2):187. https://doi.org/10.3390/metabo12020187
Chicago/Turabian StyleBeger, Aaron W., Beatrix Dudzik, Randall L. Woltjer, and Paul L. Wood. 2022. "Human Brain Lipidomics: Pilot Analysis of the Basal Ganglia Sphingolipidome in Parkinson’s Disease and Lewy Body Disease" Metabolites 12, no. 2: 187. https://doi.org/10.3390/metabo12020187
APA StyleBeger, A. W., Dudzik, B., Woltjer, R. L., & Wood, P. L. (2022). Human Brain Lipidomics: Pilot Analysis of the Basal Ganglia Sphingolipidome in Parkinson’s Disease and Lewy Body Disease. Metabolites, 12(2), 187. https://doi.org/10.3390/metabo12020187