
JPA: Joint Metabolic Feature Extraction Increases the Depth of Chemical Coverage for LC-MS-Based Metabolomics and Exposomics

 , ,
, , 
Abstract
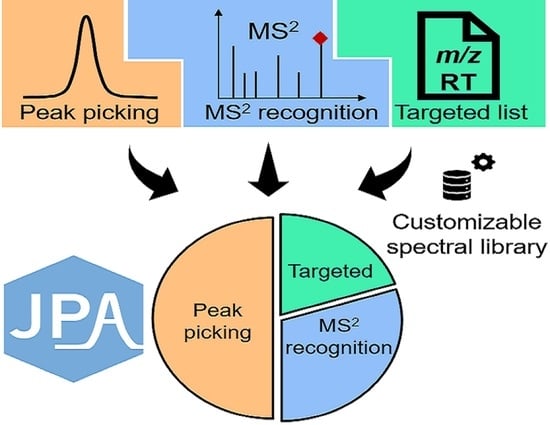
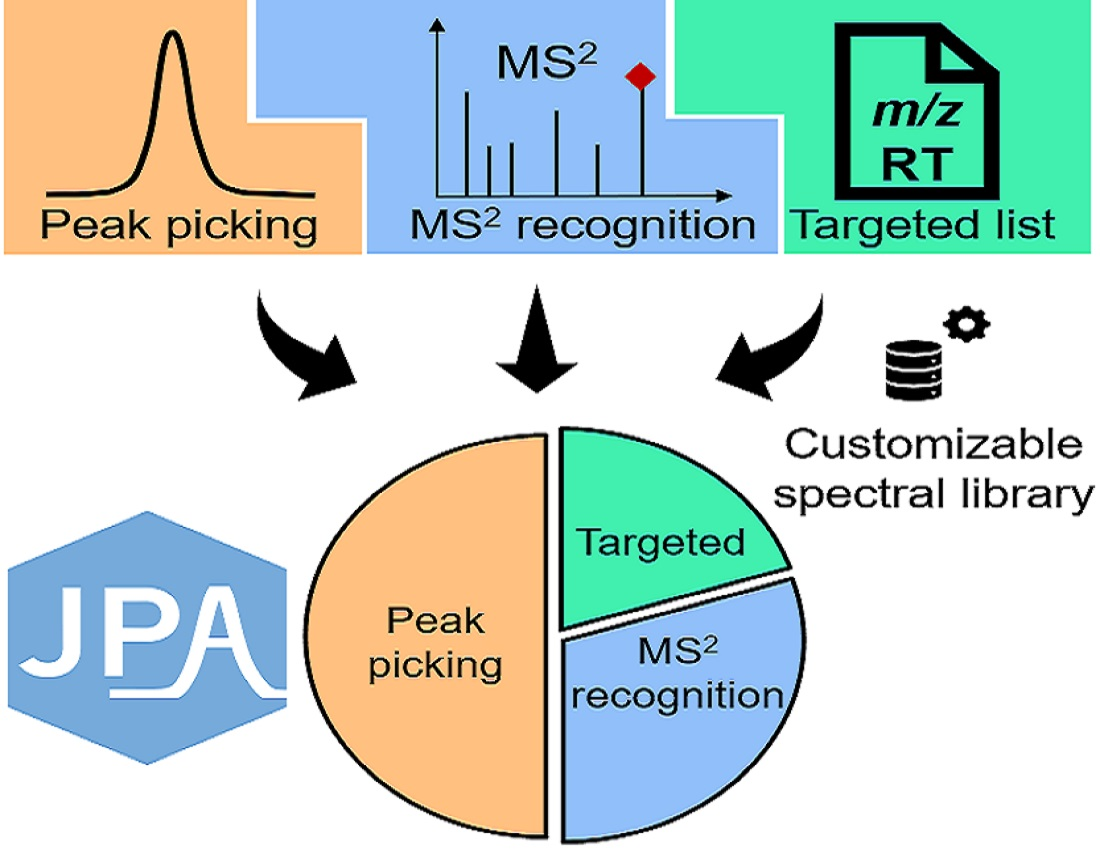
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
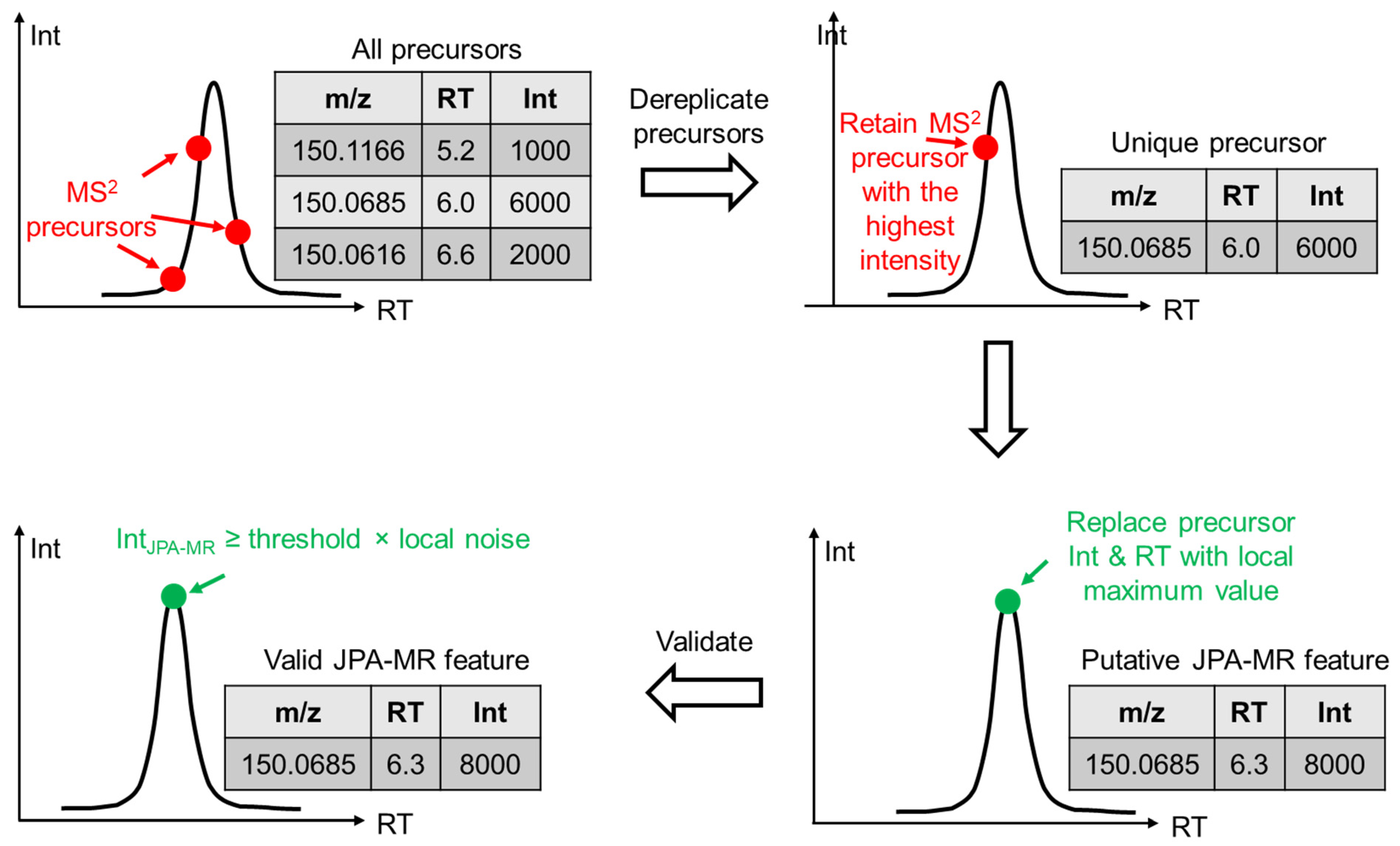
2.1. JPA Algorithms of Feature Extraction, Alignment, and Metabolite Annotation
2.2. JPA Rescues Low-Abundant Metabolic Features
2.3. Confidence of the Rescued Metabolic Features
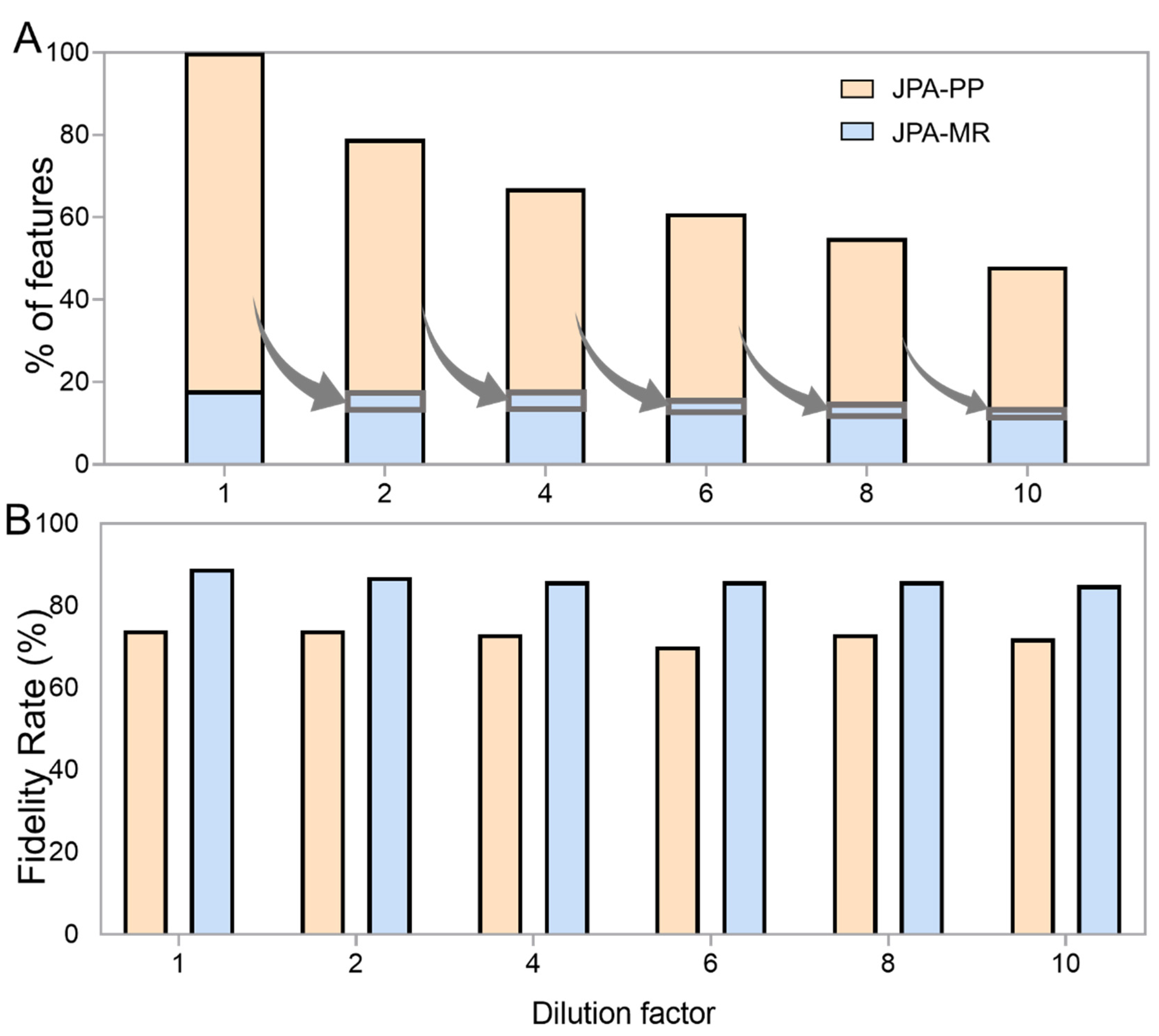
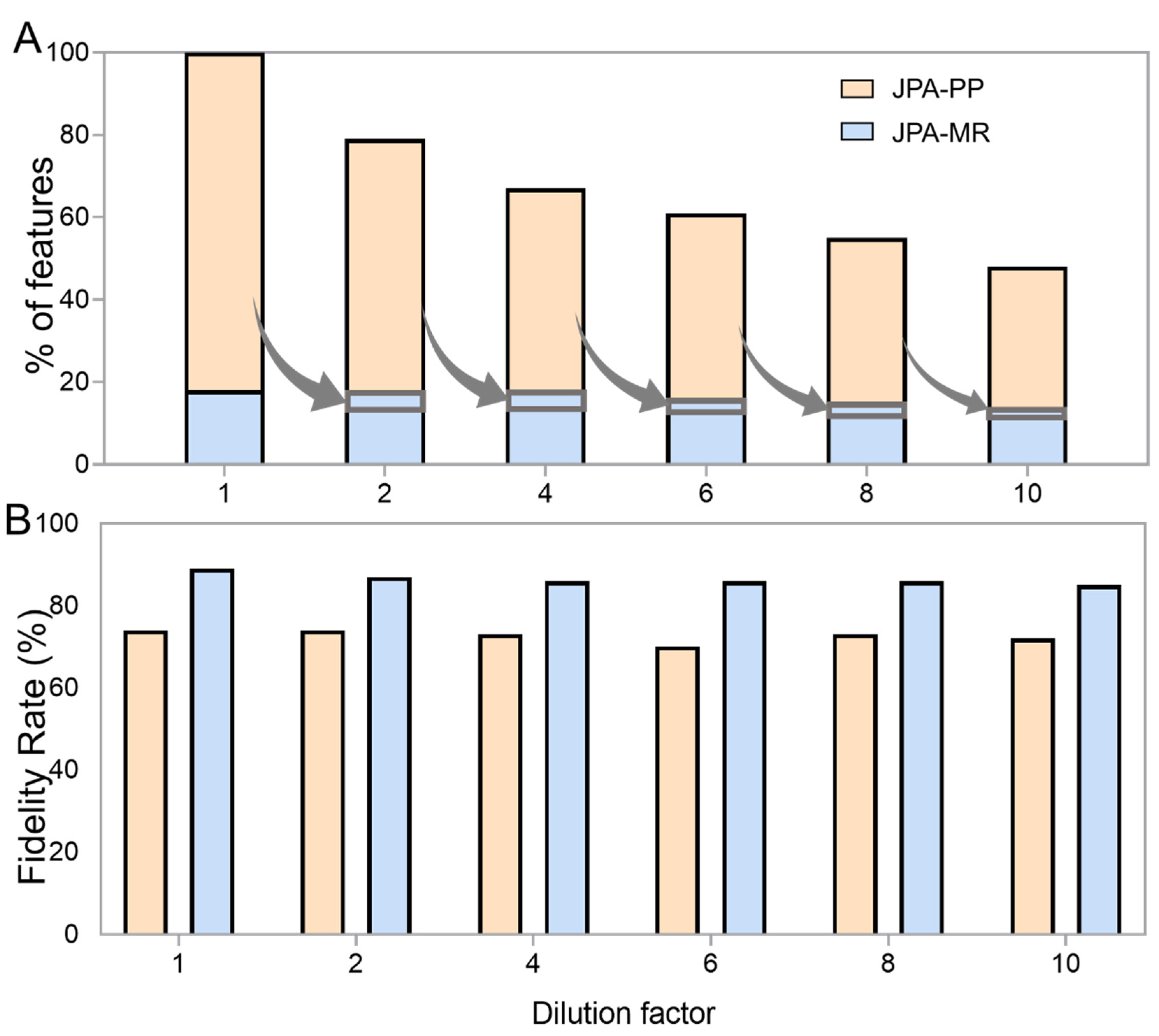
2.4. Robustness
2.5. JPA Offers Higher Analytical Sensitivity
2.6. JPA in Exposome Research
3. Materials and Methods
3.1. Metabolomics Experiments
3.2. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huan, T.; Forsberg, E.M.; Rinehart, D.; Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Fang, M.; Aisporna, A.; Hilmers, B.; Poole, F.L. Systems biology guided by XCMS Online metabolomics. Nat. Methods 2017, 14, 461–462. [Google Scholar] [CrossRef]
- Huan, T.; Palermo, A.; Ivanisevic, J.; Rinehart, D.; Edler, D.; Phommavongsay, T.; Benton, H.P.; Guijas, C.; Domingo-Almenara, X.; Warth, B. Autonomous multimodal metabolomics data integration for comprehensive pathway analysis and systems biology. Anal. Chem. 2018, 90, 8396–8403. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, C.P. The exposome: From concept to utility. Int. J. Epidemiol. 2012, 41, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Chadeau-Hyam, M.; Gmuender, H.; Gulliver, J.; Herceg, Z.; Kleinjans, J.; Kogevinas, M.; Kyrtopoulos, S.; Nieuwenhuijsen, M.; Phillips, D.H. The exposome in practice: Design of the EXPOsOMICS project. Int. J. Hyg. Environ. Health 2017, 220, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Raffeiner, P.; Granados, A.; Huan, T.; Fang, M.; Forsberg, E.M.; Benton, H.P.; Goetz, L.; Johnson, C.H.; Siuzdak, G. Metabolomics reveals that dietary xenoestrogens alter cellular metabolism induced by palbociclib/letrozole combination cancer therapy. Cell Chem. Biol. 2018, 25, 291–300.e3. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Huan, T. Comparison of full-scan, data-dependent, and data-independent acquisition modes in liquid chromatography–mass spectrometry based untargeted metabolomics. Anal. Chem. 2020, 92, 8072–8080. [Google Scholar] [CrossRef]
- Guo, J.; Huan, T. Evaluation of significant features discovered from different data acquisition modes in mass spectrometry-based untargeted metabolomics. Anal. Chim. Acta 2020, 1137, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Benton, H.P.; Ivanisevic, J.; Mahieu, N.G.; Kurczy, M.E.; Johnson, C.H.; Franco, L.; Rinehart, D.; Valentine, E.; Gowda, H.; Ubhi, B.K. Autonomous metabolomics for rapid metabolite identification in global profiling. Anal. Chem. 2015, 87, 884–891. [Google Scholar] [CrossRef] [Green Version]
- Tautenhahn, R.; Böttcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Treviño, V.; Yañez-Garza, I.L.; Rodriguez-López, C.E.; Urrea-López, R.; Garza-Rodriguez, M.L.; Barrera-Saldaña, H.A.; Tamez-Peña, J.G.; Winkler, R.; Díaz de-la-Garza, R.I. GridMass: A fast two-dimensional feature detection method for LC/MS. J. Mass Spectrom. 2015, 50, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Kanazawa, M.; Ogiwara, A.; Arita, M. MRMPROBS suite for metabolomics using large-scale MRM assays. Bioinformatics 2014, 30, 2379–2380. [Google Scholar] [CrossRef] [Green Version]
- Andreev, V.P.; Rejtar, T.; Chen, H.-S.; Moskovets, E.V.; Ivanov, A.R.; Karger, B.L. A universal denoising and peak picking algorithm for LC−MS based on matched filtration in the chromatographic time domain. Anal. Chem. 2003, 75, 6314–6326. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Cai, B.; Huan, T. Enhancing metabolome coverage in data-dependent LC–MS/MS analysis through an integrated feature extraction strategy. Anal. Chem. 2019, 91, 14433–14441. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, A.M.; Correia, G.D.S.; Sands, C.J.; Camuzeaux, S.; Yuen, A.H.Y.; Chekmeneva, E.; Takáts, Z.; Pearce, J.T.M.; Lewis, M.R. peakPantheR, an R package for large-scale targeted extraction and integration of annotated metabolic features in LC–MS profiling datasets. Bioinformatics 2021, 37, 4886–4888. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Forsberg, E.M.; Huan, T.; Rinehart, D.; Benton, H.P.; Warth, B.; Hilmers, B.; Siuzdak, G. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat. Protoc. 2018, 13, 633. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Bottcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Myers, O.D.; Sumner, S.J.; Li, S.; Barnes, S.; Du, X. Detailed investigation and comparison of the XCMS and MZmine 2 chromatogram construction and chromatographic peak detection methods for preprocessing mass spectrometry metabolomics data. Anal. Chem. 2017, 89, 8689–8695. [Google Scholar] [CrossRef]
- Go, Y.-M.; Walker, D.I.; Liang, Y.; Uppal, K.; Soltow, Q.A.; Tran, V.; Strobel, F.; Quyyumi, A.A.; Ziegler, T.R.; Pennell, K.D. Reference standardization for mass spectrometry and high-resolution metabolomics applications to exposome research. Toxicol. Sci. 2015, 148, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Vollmar, A.K.R.; Johnson, C.H. Analyzing metabolomics data for environmental health and exposome research. In Computational Methods and Data Analysis for Metabolomics; Springer: Berlin/Heidelberg, Germany, 2020; pp. 447–467. [Google Scholar]
- Jia, S.; Xu, T.; Huan, T.; Chong, M.; Liu, M.; Fang, W.; Fang, M. Chemical isotope labeling exposome (CIL-EXPOSOME): One high-throughput platform for human urinary global exposome characterization. Environ. Sci. Technol. 2019, 53, 5445–5453. [Google Scholar] [CrossRef]
- Warth, B.; Spangler, S.; Fang, M.; Johnson, C.H.; Forsberg, E.M.; Granados, A.; Martin, R.L.; Domingo-Almenara, X.; Huan, T.; Rinehart, D. Exposome-scale investigations guided by global metabolomics, pathway analysis, and cognitive computing. Anal. Chem. 2017, 89, 11505–11513. [Google Scholar] [CrossRef]
- Zhao, F.; Li, L.; Chen, Y.; Huang, Y.; Keerthisinghe, T.P.; Chow, A.; Dong, T.; Jia, S.; Xing, S.; Warth, B. Risk-Based Chemical Ranking and Generating a Prioritized Human Exposome Database. Environ. Health Perspect. 2021, 129, 47014. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Ivanisevic, J.; Benton, H.P.; Johnson, C.H.; Patti, G.J.; Hoang, L.T.; Uritboonthai, W.; Kurczy, M.E.; Siuzdak, G. Thermal degradation of small molecules: A global metabolomic investigation. Anal. Chem. 2015, 87, 10935–10941. [Google Scholar] [CrossRef] [PubMed]
- Beyer, B.A.; Fang, M.; Sadrian, B.; Montenegro-Burke, J.R.; Plaisted, W.C.; Kok, B.P.; Saez, E.; Kondo, T.; Siuzdak, G.; Lairson, L.L. Metabolomics-based discovery of a metabolite that enhances oligodendrocyte maturation. Nat. Chem. Biol. 2018, 14, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Shen, S.; Xing, S.; Chen, Y.; Chen, F.; Porter, E.M.; Yu, H.; Huan, T. EVA: Evaluation of Metabolic Feature Fidelity Using a Deep Learning Model Trained With Over 25000 Extracted Ion Chromatograms. Anal. Chem. 2021, 93, 12181–12186. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, J.; Shen, S.; Liu, M.; Wang, C.; Low, B.; Chen, Y.; Hu, Y.; Xing, S.; Yu, H.; Gao, Y.; et al. JPA: Joint Metabolic Feature Extraction Increases the Depth of Chemical Coverage for LC-MS-Based Metabolomics and Exposomics. Metabolites 2022, 12, 212. https://doi.org/10.3390/metabo12030212
Guo J, Shen S, Liu M, Wang C, Low B, Chen Y, Hu Y, Xing S, Yu H, Gao Y, et al. JPA: Joint Metabolic Feature Extraction Increases the Depth of Chemical Coverage for LC-MS-Based Metabolomics and Exposomics. Metabolites. 2022; 12(3):212. https://doi.org/10.3390/metabo12030212
Chicago/Turabian StyleGuo, Jian, Sam Shen, Min Liu, Chenjingyi Wang, Brian Low, Ying Chen, Yaxi Hu, Shipei Xing, Huaxu Yu, Yu Gao, and et al. 2022. "JPA: Joint Metabolic Feature Extraction Increases the Depth of Chemical Coverage for LC-MS-Based Metabolomics and Exposomics" Metabolites 12, no. 3: 212. https://doi.org/10.3390/metabo12030212
APA StyleGuo, J., Shen, S., Liu, M., Wang, C., Low, B., Chen, Y., Hu, Y., Xing, S., Yu, H., Gao, Y., Fang, M., & Huan, T. (2022). JPA: Joint Metabolic Feature Extraction Increases the Depth of Chemical Coverage for LC-MS-Based Metabolomics and Exposomics. Metabolites, 12(3), 212. https://doi.org/10.3390/metabo12030212