
Targeting Arginine in COVID-19-Induced Immunopathology and Vasculopathy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
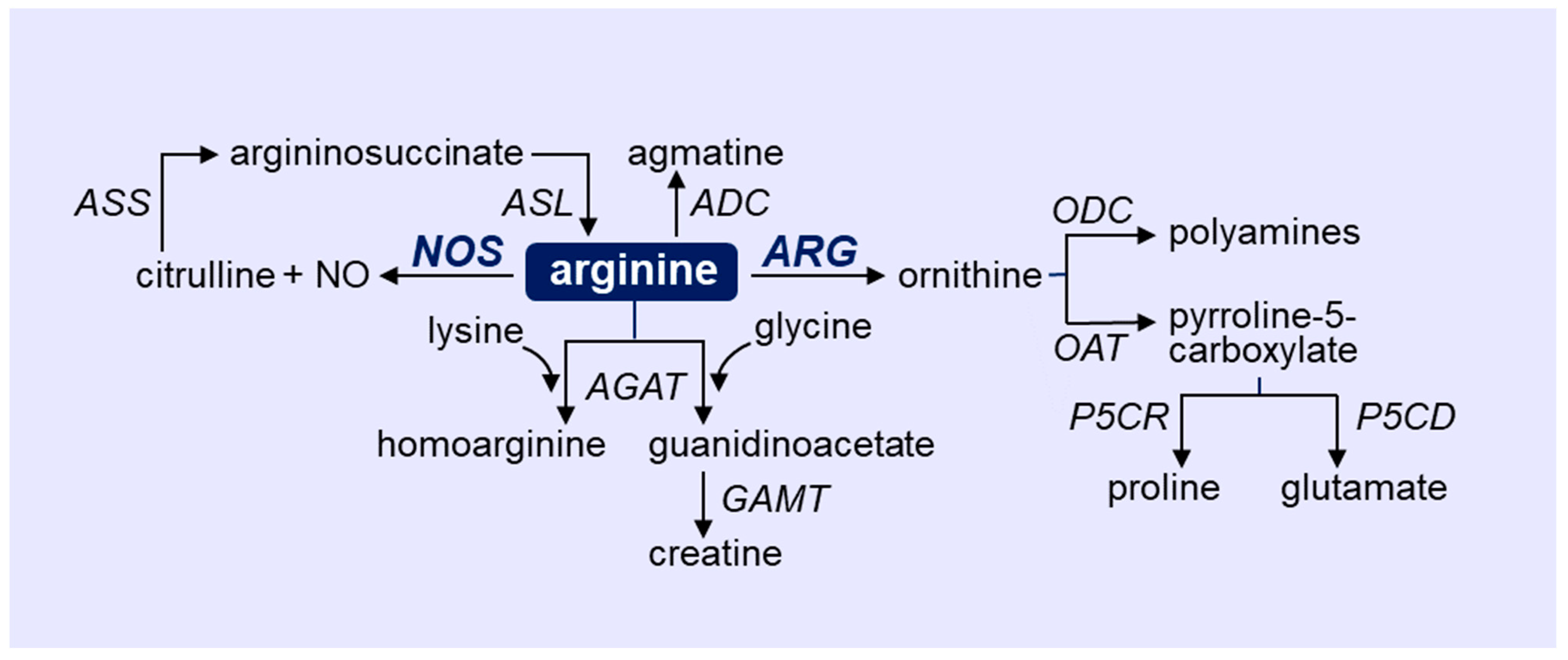
2. Overview of Arginine Metabolism
3. Role of NOS and ARG in Immune Cells
4. Role of NOS and ARG in Vascular Cells
5. Vascular Complications in COVID-19
6. Role of NOS and ARG in COVID-19
7. Targeting Arginine in COVID-19
8. Conclusions
Funding
Conflicts of Interest
References
- World Health Organization. WHO Coronavirus Disease (COVID) Dashboard. 2022. Available online: http://covid19.who.int (accessed on 21 February 2020).
- Mukra, R.; Krishan, K.; Kanchan, T. Possible modes of transmission of novel coronavirus SARS-COVID-2: A review. Acta Biomed. 2020, 91, e2020036. [Google Scholar]
- Berlin, D.A.; Gulick, R.M.; Martinez, F.J. Severe COVID-19. N. Engl. J. Med. 2020, 383, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Gautret, P.; Million, M.; Jarrot, P.A.; Camion-Jau, L.; Colson, P.; Fenollar, F.; Leone, M.; la Scola, B.; Devaux, C.; Gaubert, J.Y.; et al. Natural history of COVID-19 and therapeutic options. Expert Rev. Clin. Immunol. 2020, 16, 1159–1184. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.A. What is a host? Attributes of individual susceptibility. Infect Immun. 2018, 86, e00636-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadyen, J.D.; Stevens, H.; Peter, K. The emerging threat of (micro) thrombosis in COVID-19 and its therapeutic implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Ochoa, A.C.; Al-Khami, A.A. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front. Immunol. 2017, 8, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halaby, M.J.; McGaha, T.L. Amino acid transport and metabolism in myeloid function. Front. Immunol. 2021, 12, 695238. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ming, X.-F. Arginase: The emerging therapeutic target for vascular oxidative stress and inflammation. Front. Immunol. 2013, 4, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharm. 2009, 158, 638–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marti I Lindez, A.-A.; Reith, W. Arginine-dependent immune responses. Cell Mol. Life Sci. 2021, 78, 5303–5324. [Google Scholar] [CrossRef]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1988, 336, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Regulation of L-arginine transport and metabolism in vascular smooth muscle cells. Cell Biochem. Biophys. 2001, 35, 19–34. [Google Scholar] [CrossRef]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Arginase: A critical regulator of nitric oxide synthesis and vascular function. Clin. Exp. Pharmacol. Physiol. 2007, 34, 906–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durante, W. Role of arginase in vessel wall remodeling. Front. Immunol. 2013, 4, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Ming, X.F. Endothelial arginase: A new target in atherosclerosis. Curr. Hypertens. Rep. 2006, 8, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: Reversal of arginine steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A multifaceted enzyme important in health and disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falck-Jones, S.; Vangeti, S.; Yu, M.; Falck-Jones, R.; Cagigi, A.; Badolati, I.; Österberg, B.; Lautenbach, M.J.; Åhlberg, E.; Lin, A.; et al. Functional monocytic myeloid-derived suppressor cells increase blood but not airways and predict COVID-19 severity. J. Clin. Investig. 2021, 131, e144734. [Google Scholar] [CrossRef] [PubMed]
- Husson, A.; Brasse-Lagnel, C.; Fairand, A.; Renouf, S.; Lavoinne, A. Argininosuccinate synthetase for the urea cycle to the citrulline-NO cycle. Eur. J. Biochem. 2003, 270, 1887–1899. [Google Scholar] [CrossRef] [PubMed]
- Fostermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Jenkinson, C.P.; Grody, W.W.; Cederbaum, S.D. Comparative properties of arginase. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1996, 114B, 107–132. [Google Scholar] [CrossRef]
- Tabor, C.W.; Tabor, H. Polyamines. Ann. Rev. Biochem. 1984, 53, 749–790. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Liao, L.; Peyton, K.J.; Schafer, A.I. Thrombin stimulates vascular smooth muscle cell polyamine synthesis by inducing cationic amino acid transporter and ornithine decarboxylase activity. Circ. Res. 1998, 83, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Liao, L.; Reyna, S.V.; Peyton, K.J.; Schafer, A.I. Physiologic cyclic stretch directs L-arginine transport and metabolism to collagen synthesis in vascular smooth muscle cells. FASEB J. 2000, 14, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Liao, L.; Reyna, S.V.; Peyton, K.J.; Schafer, A.I. Transforming growth factor-β1 stimulates L-arginine transport and metabolism in vascular smooth muscle cells: Role in polyamine and collagen synthesis. Circulation 2001, 103, 1121–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Bugaj, L.J.; Oh, Y.J.; Bivalacqua, T.J.; Ryhoo, S.; Soucy, K.G.; Santhanam, L.; Webb, A.; Camara, A.; Sikka, G.; et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J. Appl. Physiol. 2009, 107, 1249–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossner, J.; Hammermann, R.; Racke, K. Concomitant down-regulation of L-arginine transport and nitric oxide (NO) synthesis in rat alveolar macrophages by the polyamine spermine. Pulm. Pharmacol. Ther. 2001, 14, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Rhu, H.; Ferrante, R.J.; Morris, S.M.; Ratan, R.R. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc. Natl. Acad. Sci. USA 2003, 100, 4843–4848. [Google Scholar] [CrossRef] [Green Version]
- Daghigh, F.; Fukuto, J.M.; Ash, D.E. Inhibition of rat liver arginase by an intermediate in NO biosynthesis, NG-hydroxy-L-arginine: Implications for the regulation of nitric oxide biosynthesis by arginase. Biochem. Biophys. Res. Commun 1994, 202, 174–180. [Google Scholar] [CrossRef]
- Santhanam, L.; Lim, H.K.; Miriel, V.; Brown, T.; Patel, M.; Balanson, S.; Ryoo, S.; Anderson, M.; Irani, K.; Khanday, F.; et al. Inducible NO synthase-dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ. Res. 2007, 101, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Karupiah, G.; Harris, N. Inhibition of viral replication by nitric oxide and its reversal by ferrous sulfate and tricarboxylic acid cycle intermediates. J. Exp. Med. 1995, 181, 2171–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanishi, M. Nitric oxide inhibits Epstein-Barr virus DNA replication and activation of latent EBV. Intervirology 1995, 38, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.; Buller, R.M.; Karupiah, G. Gamma interferon-induced nitric oxide-mediated inhibition of vaccinia virus replication. J. Virol. 1995, 69, 910–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowenstein, C.J.; Hill, S.L.; Lafond-Walker, A.; Wu, J.; Allen, G.; Landavere, M.; Rose, N.R.; Herskowitz, A. Nitric oxide inhibits viral replication in murine myocarditis. J. Clin. Investig. 1996, 97, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, M.; Zaragoza, C.; McMillan, A.; Quick, R.A.; Hohenadl, C.; Lowenstein, J.M.; Lowenstein, C.J. An antiviral mechanism of nitric oxide: Inhibition of a viral protease. Immunity 1999, 10, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Akerstrom, S.; Mousavi-Jazi, M.; Klingstrom, J.; Leijon, M.; Lundkvist, A.; Mirazimi, A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1966–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerstrom, S.; Gunalan, V.; Keng, C.T.; Tan, Y.-J.; Mirazimi, A. Dual effect of nitric oxide on SARS-CoV replication: Viral RNA production and palmitoylation of the S protein are affected. Virology 2009, 395, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Louis, C.A.; Mody, V.; Henry, W.L.; Reichner, J.S.; Albina, J.E. Regulation of arginase isoforms I and II by IL-4 in cultured murine peritoneal macrophages. Am. J. Physiol. 1999, 276, R237–R242. [Google Scholar] [CrossRef] [Green Version]
- Albina, J.E.; Mills, C.D.; Barbul, A.; Thirkill, C.E.; Henry, W.L., Jr.; Mastrofrancesco, B.; Caldwell, M.D. Arginine metabolism in wounds. Am. J. Physiol. 1988, 254, E459–E467. [Google Scholar] [CrossRef] [PubMed]
- Dunand-Sauthier, I.; Irla, M.; Carnesechi, S.; Seguin-Estevez, Q.; Vejnar, C.E.; Zdobnov, E.M.; Santiago-Raber, M.-L.; Reith, W. Repression of arginase-2 expression in dendritic cells by microRNA-155 is critical for promoting T cell proliferation. J. Immunol. 2014, 193, 1690–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munder, M.; Schneider, H.; Luckner, C.; Giese, T.; Langhans, C.D.; Fuentes, J.M.; Kropf, P.; Mueller, I.; Kolb, A.; Modolell, M.; et al. Suppression of T cell functions by granulocyte arginase. Blood 2006, 108, 1627–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotondo, R.; Bertolotto, M.; Barisione, G.; Astigiano, S.; Mandruzzato, S.; Ottonello, L.; Dallegri, F.; Bronte, V.; Ferrini, S.; Barbieri, O. Exocytosis of azurophil and arginase-1-containing granules by activated polymorphonuclear neutrophils is required to inhibit T lymphocyte proliferation. J. Leukoc. Biol. 2011, 89, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell receptor responses. Cancer Res. 2004, 64, 5838–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zea, A.H.; Rodriguez, P.C.; Culotta, K.S.; Hernandez, C.P.; DeSalvo, J.; Ochoa, J.B.; Park, H.; Zabaleta, J.; Ochoa, A.C. L-Arginine modulates CD3zeta expression and T cell function in activated T lymphocytes. Cell. Immunol. 2004, 232, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. l-Arginine-depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taheri, F.; Ochao, J.B.; Faghiri, Z.; Culotta, K.; Park, H.J.; Lan, M.S.; Zea, A.H.; Ochoa, A.C. L-Arginine regulates the expression of the T-cell receptor zeta chain (CD3zeta) in Jurkat cells. Clin. Cancer Res. 2001, 7, 958s–965s. [Google Scholar] [PubMed]
- Rodriguez, P.C.; Hernandez, C.P.; Morrow, K.; Sierra, R.; Zabaleta, J.; Wyczechowska, D.D.; Ochoa, A.C. L-Arginine deprivation regulates cyclin D3 mRNA stability in human T cells by controlling HuR expression. J. Immunol. 2010, 185, 5198–5204. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-Arginine availability regulates T-lymphocyte cell cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Velde, L.A.; Murray, P.J. Proliferating helper T cells require Rictor/mTORC2 complex to integrate signals from limiting environmental amino acids. J. Biol. Chem. 2016, 291, 25815–25822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzoni, A.; Bronte, V.; Visintin, A.; Spitzer, J.H.; Apolloni, E.; Serafini, P.; Zanovello, P.; Segal, D.M. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J. Immunol. 2002, 168, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Saio, M.; Radoja, S.; Marino, M.; Frey, A.B. Tumor-infiltrating macrophages induce apoptosis in activated CD8(+) T cells by a mechanism requiring cell contact and mediated by both the cell-associated form of TNF and nitric oxide. J. Immunol. 2001, 167, 5583–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peranzoni, E.; Marigo, I.; Dolcetti, L.; Ugel, S.; Sonda, N.; Taschin, E.; Mantelli, B.; Bronte, V.; Zanovello, P. Role of arginine metabolism in immunity and immunopathology. Immunobiol 2007, 212, 795–812. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Srivastava, S.; Kandal, U.; Sade, H.; Lewis, V.; Sarin, A.; George, A.; Bal, V.; Durdik, J.M.; Rath, S.; et al. Inducible nitric oxide synthase in T cells regulates T cell death and immune memory. J. Clin. Investig. 2004, 113, 1734–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, W.J.; Kwikkers, K.L.; te Velde, A.A.; van Deventer, S.J.H.; Nolte, M.A.; Mebius, R.E.; Ruijter, J.M.; Lamers, M.C.; Lamers, W.H. Arginine deficiency affects early B cell maturation and lymphoid organ development in transgenic mice. J. Clin. Investig. 2002, 110, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.S.; Shenoy, G.N.; Rath, S.; Bal, V.; George, A. Inducible nitric oxide synthase is a major intermediate in signaling pathways for the survival of plasma cells. Nat. Immunol. 2014, 15, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Park, K.G.; Hayes, P.D.; Garlick, P.J.; Sewell, H.; Eremin, O. Stimulation of lymphocyte natural cytotoxicity by L-arginine. Lancet 1991, 337, 645–646. [Google Scholar] [CrossRef]
- Brittenden, J.; Park, K.G.; Heys, S.D.; Ross, C.; Ashby, J.; Ah-See, A.K.; Eremin, O. L-arginine stimulates host defenses in patients with breast cancer. Surgery 1994, 115, 205–212. [Google Scholar] [PubMed]
- Reynolds, J.V.; Daly, J.M.; Zhang, S.; Evantash, E.; Shou, J.; Sigal, R.; Ziegler, M.M. Immunomodulatory mechanisms of arginine. Surgery 1988, 104, 142–151. [Google Scholar] [PubMed]
- Lamas, B.; Vergnaud-Gauduchon, J.; Goncalves-Mendes, N.; Perche, O.; Rossary, A.; Vasson, M.-P.; Farges, M.-C. Altered functions of natural killer cells in response to L-arginine availability. Cell. Immunol. 2012, 280, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Faruke, K.; Burd, P.R.; Horvath-Arcidicono, J.A.; Hori, K.; Mostowski, H.; Bloom, E.T. Human NK cells express endothelial nitric oxide synthase, and nitric oxide protects them from activation-induced cells death by regulating expression of TNF-alpha. J. Immunol. 1999, 163, 1473–1480. [Google Scholar]
- Jyothi, M.D.; Khar, A. Interleukin-2-induced nitric oxide synthase and nuclear factor-kappaB activity in activated natural killer cells and the production of interferon-gamma. Scand. J. Immunol. 2000, 52, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Oberlies, J.; Watzl, C.; Giese, A.T.; Luckner, C.; Kropf, P.; Muller, I.; Ho, A.D.; Munder, M. Regulation of NK cell function by human granulocyte arginase. J. Immunol. 2009, 182, 5259–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, C.C.; Roggerson, K.M.; Lee, H.-C.; Golden-Mason, L.; Rosen, H.R.; Hahn, Y.S. Hepatitits C virus-induced myeloid-derived suppressor cells suppress NK cell IFN-γ production by altering cellular metabolism via arginase-1. J. Immunol. 2016, 196, 2283–2292. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Arora, R.C.; Hiebert, B.M.; Lerner, B.; Szwajcer, A.; McDonald, K.; Rigatto, C.; Komenda, P.; Sood, M.; Tangri, N. Non-invasive endothelial function testing and the risk of adverse outcomes: A systematic review and meta-analysis. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 736–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, J.O.; Gladwin, M.T.; Weitzberg, E. Strategies to increase nitric oxide signalling in cardiovascular disease. Nat. Rev. Drug. Discov. 2015, 14, 623–641. [Google Scholar] [CrossRef] [PubMed]
- Elms, S.; Chen, F.; Wang, Y.; Qian, J.; Askari, B.; Yu, Y.; Pandey, D.; Iddings, J.; Caldwell, R.B.; Fulton, D.J.R. Insights into the arginine paradox: Evidence against the importance of subcellular localization of arginase and eNOS. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H651–H666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdi, A.; Kovames, O.; Pernow, J. Improvement in endothelial function in cardiovascular disease—is arginase the target? Int. J. Cardiol. 2020, 301, 207–214. [Google Scholar] [CrossRef]
- Zhu, W.; Chandrsekharan, U.M.; Bandyopadhyay, S.; Morris, S.M., Jr.; DiCorleto, P.E.; Kashyap, V.S. Thrombin induces endothelial arginase through AP-1 activation. Am. J. Physiol. Cell Physiol. 2010, 298, C952–C960. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Zhu, W.; Pavkov, M.L.; Kinney, C.M.; DiCorleto, P.E.; Kashyap, V.S. Arginase blockade lessens endothelial dysfunction after thrombosis. Vasc. Surg. 2008, 48, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, S.; Gupta, G.; Benjo, A.; Lim, H.K.; Camara, A.; Sikkha, G.; Lim, H.K.; Sohi, J.; Santhanam, L.; Soucy, K.; et al. Endothelial arginase II: A novel target for the treatment of atherosclerosis. Circ. Res. 2008, 102, 923–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, F.K.; Peyton, K.J.; Liu, X.M.; Azam, M.A.; Shebib, A.R.; Johnson, R.A.; William, D. Arginase promotes endothelial dysfunction and hypertension in obese rats. Obesity 2015, 23, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, F.K.; Johnson, R.A.; Peyton, K.J.; Shebib, A.R.; Durante, W. Arginase promotes skeletal muscle arteriolar endothelial dysfunction in diabetic rats. Front. Immunol. 2013, 4, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, M.J.; Platt, D.H.; Yawfik, H.E.; Labazi, M.; El-Remessy, A.B.; Bartoli, M.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ. Res. 2008, 102, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.K.; Johnson, R.A.; Peyton, K.J.; Durante, W. Arginase inhibition restores arterial endothelial dysfunction in Dahl rats with salt-induced hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1057–R1062. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.K.; Durante, W.; Craig, T.; Peyton, K.J.; Myers, J.G.; Stewart, R.M. Vascular arginase contributes to arteriolar endothelial dysfunction in a rat model of hemorrhagic shock. J. Trauma. 2010, 69, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Gonon, A.T.; Sjoquist, P.O.; Lundberg, J.O.; Pernow, J. Arginase inhibition mediates cardioprotection during ischemia-reperfusion. Cardiovasc. Res. 2010, 85, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppan, J.; Tran, H.T.; Bead, V.R.; Oh, Y.J.; Sikka, G.; Bivalacqua, T.J.; Burnett, A.L.; Berkowitz, D.E.; Santhanam, L. Arginase inhibition reverses endothelial dysfunction, pulmonary hypertension, and vascular stiffness in transgenic sickle cell mice. Anesth. Analg. 2016, 123, 652–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demougeot, C.; Prigent-Tessier, A.; Marie, C.; Berthelot, A. Arginase inhibition reduced endothelial dysfunction and blood pressure rising in spontaneously hypertensive rats. J. Hypertens. 2005, 23, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.; Lee, C.; Kang, M.; Huang, Y.; Giordano, F.J.; Lee, P.J.; Trow, T.K.; Homer, R.J.; Sessa, W.C.; Elias, J.A.; et al. IL-13 receptor α2-arginase 2 pathway mediates IL-13-iinduced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L112–L124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.H.; Wu, G.; Morris, S.M., Jr.; Ignarro, L.J. Elevated arginase 1 expression in rat aortic smooth muscle cells increases cell proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 9260–9264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyton, K.J.; Ensenat, D.; Azam, M.A.; Keswani, A.N.; Kanna, S.; Liu, X.-M.; Wang, H.; Tulis, D.A.; Durante, W. Arginase promotes neointima formation in rat injured carotid arteries. Arter. Thromb. Vasc. Biol. 2009, 29, 488–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnost, T.; Ma, L.; da Silva, R.F.; Rezakhaniha, R.; Houdayer, C.; Stergiopulos, N.; André, C.; Guillaume, Y.; Berthelot, A.; Demougeot, C. Cardiovascular effects of arginase inhibition in spontaneously hypertensive rats with fully developed hypertension. Cardiovasc. Res. 2010, 87, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatta, Y.; Yao, L.; Haque, H.A.; Shatanawi, A.; Xu, Z.; Caldwell, R.B.; Caldwell, R.W. Angiotensin II-induced arterial thickening, fibrosis and stiffening involve elevated arginase function. PLoS ONE 2015, 10, e0121727. [Google Scholar]
- Grasemann, H.; Dhaliwal, R.; Ivanovska, J.; Kantores, C.; McNamara, P.J.; Scott, J.A.; Belik, J.; Jankov, R.P. Arginase inhibition prevents bleomycin-induced pulmonary vascular remodeling, and collagen deposition in neonatal rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L503–L510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaco, R.D.; Southwood, M.; Toshner, M.; Alexander, L.E.C.; Morrell, N.; Chilvers, E.; et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatta, A.; Yao, L.; Xu, Z.; Toque, H.A.; Chen, J.; Atawia, R.T.; Fouda, A.Y.; Bagi, Z.; Lucas, R.; Caldwell, R.B.; et al. Obesity-induced vascular dysfunction and arterial stiffening requires endothelial arginase 1. Cardiovasc. Res. 2017, 113, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, D.; Sperhake, J.P.; Lutgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy findings and venous thromboembolism in patients with COVID-19: A prospective cohort study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Q.; Wang, Y.; Wu, Y.; Xu, J.; Yu, Y.; Shange, Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J. Thromb. Haemostas 2020, 18, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Favoloro, E.J. D-dimer is associated with severity of coronavirus disease 2019: A pooled analysis. J. Thromb. Haemostas. 2020, 18, 876–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippi, G.; Phlebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chem. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Modin, D.; Claggett, B.; Sindet-Pedersen, C.; Lassen, M.C.H.; Skaarup, K.G.; Jensen, J.U.S.; Fralick, M.; Schou, M.; Lamberts, M.; Gerds, T.; et al. Acute COVID-19 and the incidence of ischemic stroke and acute myocardial infarction. Circulation 2020, 142, 2080–2082. [Google Scholar] [CrossRef] [PubMed]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-vessel stroke as a presenting feature of COVID-19 in the young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Urquijo, M.; Gonzalez-Rayes, J.M.; Castro-Varela, A.; Hinojosa-Gonzalez, D.E.; Ramos-Cazares, R.E.; Vazquez-Garza, E.; Paredes-Vazquez, J.G.; Castillo-Perez, M.; Jerjes-Sanchez, C.; Fabiani, M.A.; et al. Unexpected arterial thrombosis and acute limb ischemia in COVID-19 patients. Results from the Ibero-Latin American acute arterial thrombosis registry in COVID-19: (ARTICO-19). Vascular 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Havervich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis, in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhary, I.; Fink, S.L.; Xu, H.; Najafian, B.; Deutsch, G.; Lacy, J.M.; Williams, T.; et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: A case series. Lancet 2020, 396, 320–332. [Google Scholar] [CrossRef]
- Gattinoni, L.; Chiumello, D.; Caironi, P.; Busana, M.; Romitti, F.; Brazzi, L.; Camporota, L. COVID-19 pneumonia: Different respiratory treatments for different phenotypes? Intensive Care Med. 2020, 46, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.; Charytan, D.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinical 2020, 24, 100434. [Google Scholar] [CrossRef] [PubMed]
- Viola, F.; Pignatelli, P.; Cammisotto, V.; Bartimoccia, S.; Carnevale, R.; Nocella, C. COVID-19 and thrombosis: Clinical features, mechanism of disease, and therapeutic implications. Kardiol. Pol. 2021, 79, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endothelialiopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2021, 18, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Nicosia, R.F.; Ligresti, G.; Caporarello, N.; Akilesh, S.; Ribatti, D. COVID-19 Vasculopathy: Mounting evidence for an indirect mechanism of endothelial injury. Am. J. Pathol. 2021, 191, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yang, K.Y.; Huang, Y.; Liu, K.O. Endothelial contribution to COVID-19: An update on mechanisms and therapeutic implications. J. Mol. Cell. Cardiol. 2022, 164, 69–82. [Google Scholar] [CrossRef]
- Prasad, M.; Leion, M.; Lerman, L.O.; Lerman, A. Viral endothelial dysfunction: A unifying mechanism for COVID-19. Mayo. Clin. Proc. 2021, 96, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endothelialitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Fox, S.E.; Lameira, F.S.; Rinker, E.B.; Vander Heide, R.S. Cardiac endothelialitis and multisystem inflammatory syndrome after COVID-19. Ann. Intern. Med. 2020, 173, 1025–1027. [Google Scholar] [CrossRef]
- Carnevale, S.; Beretta, P.; Morbini, P. Direct endothelial damage and vasculitis due to SARS-CoV-2 in small bowel submucosa of CIVD-19 patients with diarrhea. J. Med. Virol. 2021, 93, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Guadagno, A.; Greco, M.; Parodi, A.; Burlando, M. An unusual case of bullous haemorrhagic vasculitis in a COVID-19 patient. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e675–e676. [Google Scholar] [CrossRef] [PubMed]
- Escher, R.; Breakey, N.; Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.D.; Patterson, E.K.; Daley, M.; Cepinskas, G.; on behalf of the Lawson COVID-19 Study Team. Case report: Inflammation and endothelial injury profiling of COVID-19 pediatric multisystem inflammatory syndrome (MIS-C). Fronti. Pediatr. 2021, 9, 597926. [Google Scholar] [CrossRef] [PubMed]
- Crippa, S.; Kagi, G.; Graf, L.; Meyer Sauteur, P.M.; Kohler, P. Stroke in young adult with mild COVID-19 suggesting endothelialitis. New Microbes New Infect. 2020, 38, 100781. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, C.; Burtey, S.; Sabatier, F.; Cauchois, R.; Lano, G.; Abdili, E.; Daviet, F.; Arnaud, L.; Brunet, P.; Hraiech, S.; et al. Circulating endothelial cells as a marker of endothelial injury in severe COVID-19. J. Infect. Dis. 2020, 222, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Neri, T.; Nieri, D.; Celi, A. P-selectin blockade in COVID-19-related ARDS. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1237–L1238. [Google Scholar] [CrossRef]
- Smadja, D.M.; Guerin, C.L.; Chocron, R.; Yatim, N.; Boussier, J.; Gendron, N.; Khider, L.; Hadjadj, J.; Goudot, G.; Debuc, B.; et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 2020, 23, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, S.; Fogagnolo, A.; Campo, G.; Zucchetti, O.; Verri, M.; Ottaviani, I.; Tunstall, T.; Grasso, S.; Scaramuzzo, V.; Murgolo, F.; et al. Markers of endothelial and epithelial pulmonary injury in mechanically-ventilated COVID-19 ICU patients. Crit. Care 2021, 25, 74. [Google Scholar] [CrossRef]
- Sabioni, L.R.; Tibirica, E.; Lamas, C.C.; Amorim, G.D.; De Lorenzo, A. Systemic microvascular dysfunction in COVID-19. Am. J. Cardiovasc. Dis. 2020, 10, 386–391. [Google Scholar]
- Sabioni, L.; De Lorenzo, A.; Lamas, C.; Muccillo, F.; Castro-Faria-Neto, H.C.; Estato, V.; Tibirica, E. Systemic microvascular endothelial dysfunction and disease severity in COVID-19 patients: Evaluation by laser doppler perfusion monitoring and cytokine/chemokine analysis. Microvasc. Res. 2021, 134, 104119. [Google Scholar] [CrossRef] [PubMed]
- Ratchford, S.M.; Stickford, J.L.; Province, V.M.; Stute, N.; Augenreich, M.A.; Koontz, L.K.; Bobo, L.K.; Stickford, A.S.L. Vascular alterations among young adults with SARS-CoV-2. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H404–H410. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, D.; Song, J.W.; Zullo, J.; Lipphardt, M.; Coneh-Gould, L.; Goligorsky, M.S. Endothelial cell dysfunction and the glycocalyx—a vicious circle. Matrix Biol. 2019, 71–72, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.D.; Patterson, E.K.; Slessarev, M.; Gill, S.E.; Martinc, C.; Daley, M.; Miller, M.R.; Patel, M.A.; Santos, C.C.D.; Bosma, K.J.; et al. Endothelial injury and glycocalyx degradation in critically ill coronavirus disease 2019 patients: Implications for microvascular platelet aggregation. Crit. Care Explor. 2020, 2, e0194. [Google Scholar] [CrossRef] [PubMed]
- Du Preez, H.N.; Aldous, C.; Hayden, M.R.; Kruger, H.G.; Lin, J. Pathogenesis of COVID-19 described through the lens of undersulfated and degraded epithelial and endothelial glycocalyx. FASEB J. 2022, 36, e22052. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Mamun, A.; Dominic, A.; Le, N.T. SARS-CoV-2 mediated endothelial dysfunction. The potential role of chronic oxidative stress. Front. Physiol. 2020, 11, 605908. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, A.; Varzideh, F.; Wilson, S.; Gambardella, J.; Eacobacci, M.; Jankauskas, S.S.; Donkor, K.; Kansakar, U.; Trimarco, V.; Mone, P.; et al. L-Arginine and COVID-19: An update. Nutrients 2021, 13, 3951. [Google Scholar] [CrossRef]
- Guimaraes, L.M.F.; Rossini, C.V.T.; Lameu, C. Implications of SARS-CoV-2 infection on eNOS and iNOS activity: Consequences for the respiratory and vascular systems. Nitric. Oxide. 2021, 111–112, 64–71. [Google Scholar] [CrossRef]
- Fang, W.; Jiang, J.; Su, L.; Shu, T.; Liu, H.; Lai, S.; Ghiladi, R.A.; Wang, J. The role of NO in COVID-19 and potential therapeutic strategies. Free Radic. Biol. Med. 2021, 163, 153–162. [Google Scholar] [CrossRef]
- Alamdari, D.H. Application of methylene blue-vitamin C-N-acetyl cysteine for treatment of critical ill COVID-19 patients, report of a phase -I clinical trial. Eur. J. Pharmacol. 2020, 885, 173494. [Google Scholar] [CrossRef]
- Derakhshani, A.; Hemmat, N.; Asadzadeh, Z.; Ghaseminia, M.; Shadbad, M.A.; Jadideslam, G.; Silvestris, N.; Racanelli, V.; Baradaran, B. Arginase 1 (Arg1) as an up-regulated gene in COVID-19 patients: A promising marker of COVID-19 immunopathy. J. Clin. Med. 2021, 10, 1050. [Google Scholar] [CrossRef] [PubMed]
- Hemmat, N.; Derakhshani, A.; Baghi, H.B.; Silvestris, N.; Baradaran, B.; De Summa, S. Neutrophils, crucial, or harmful immune cells involved in coronavirus infection: A bioinformatics study. Front. Genet. 2020, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Syrimi, E.; Fennell, E.; Richter, A.; Vrljicak, P.; Stark, R.; Ott, S.; Murray, P.G.; Al-Abadi, E.; Chikermane, A.; Dawson, P.; et al. The immune landscape of SARS-CoV-2-associated multisystem inflammatory syndrome in children (MIS-C) from acute disease to recovery. iScience 2021, 24, 103215. [Google Scholar] [CrossRef] [PubMed]
- Reizine, F.; Lesouhaitier, M.; Gregoire, M.; Pinceaux, K.; Gacouin, A.; Maamare, A.; Painvin, B.; Camus, C.; le Tulzo, Y.; Tattevin, P.; et al. SARS-CoV-2-induced ARDS associates with MDSC expansion, lymphocyte dysfunction, and arginine shortage. J. Clin. Immunol. 2021, 41, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.J.; Ochoa, J.B.; Sanchez-Pino, M.D.; Zabaleta, J.; Garai, J.; Del Valle, L.; Wyczechowska, D.; Baiamonte, L.B.; Philbrook, P.; Majumder, R.; et al. Severe COVID-19 is characterized by an impaired type I interferon response and elevated levels of arginase producing granulocytic myeloid derived suppressor cells. Front. Immunol. 2021, 12, 695972. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, A.; Grassi, G.; Notari, S.; Gili, S.; Bordoni, V.; Tartaglia, E.; Casetti, R.; Cimini, E.; Mariotti, D.; Garotto, G.; et al. Expansion of myeloid derived suppressor cells contributes to platelet activation by L-arginine deprivation during SARS-CoV-2 infection. Cells 2021, 10, 211. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Thomas, T.; Akpan, I.J.; Reisz, J.A.; Cendali, F.I.; Gamboni, F.; Nemkov, T.; Thangaraju, K.; Katneni, U.; Tanaka, K.; et al. Biological and clinical factors contributing to the metabolic heterogeneity of hospitalized patients with and without COVID-19. Cells 2021, 10, 2293. [Google Scholar] [CrossRef] [PubMed]
- She, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 2020, 182, 59–72. [Google Scholar]
- Wu, P.; Chen, D.; Ding, W.; Wu, P.; Hou, H.; Bai, Y.; Zhou, Y.; Li, K.; Xiang, S.; Liu, P.; et al. The trans-omics landscape of COVID-19. Nat. Commun. 2021, 12, 4543. [Google Scholar] [CrossRef] [PubMed]
- Rees, C.A.; Rostade, C.A.; Mantus, G.; Anderson, E.J.; Chahroudi, A.; Jaggi, P.; Wrammert, J.; Ochoa, J.B.; Ochoa, A.; Basu, R.K.; et al. Altered amino acid profile in patients with SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2101708118. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Cho, L.; Brennan, D.M.; Hazen, S.L. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J. Am. Coll. Cardiol. 2009, 53, 2061–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. J. Am. Coll. Cardiol. Basic Trans Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Meininger, C.J. Arginine nutrition and cardiovascular function. J. Nutr. 2000, 130, 2626–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durante, W. Amino acid in circulatory function and health. Adv. Exp. Med. Biol. 2020, 1265, 39–56. [Google Scholar] [PubMed]
- Grimes, J.M.; Khan, S.; Badeaux, M.; Rao, R.M.; Rowlinson, S.W.; Carvajal, R.D. Arginine depletion as a therapeutic approach for patients with COVID-19. Int. J. Infect. Dis. 2021, 102, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Melano, I.; Kuo, L.-L.; Lo, Y.-C.; Sung, P.-W.; Tien, N.; Su, W.-C. Effects of basic amino acids and their derivatives on SARS-CoV-2 and influenza-A virus infection. Viruses 2021, 13, 1301. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, G.; Coppola, A.; Izzo, R.; Annunziata, A.; Bernardo, M.; Lombardi, A.; Trimarco, V.; Santulli, G.; Trimarco, B. Effects of adding L-arginine to standard therapy with COVID-19: A randomized, double-blind, placebo-controlled, parallel-group trial. Results of the first interim analysis. EClinicalMedicine 2021, 40, 101125. [Google Scholar] [CrossRef] [PubMed]
- McNeal, C.J.; Meininger, C.J.; Wilborn, C.D.; Tekwe, C.D.; Wu, G. Safety of dietary supplementation with arginine in adult humans. Amino Acids 2018, 50, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Schwedhelm, E.; Maas, R.; Freese, R.; Jung, D.; Lukas, Z.; Jumbrecina, A.; Spickler, W.; Schulze, F.; Böger, R.H. Pharmacokinetic and pharmacodynamic properties of oral L-citrulline and L-arginine: Impact on nitric oxide metabolism. Br. J. Clin. Pharmacol. 2008, 65, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Pioggia, G.; Negrini, S. Vitamin D and COVID-19: An update on evidence and potential therapeutic implications. Clin. Mol. Allergy 2020, 18, 23. [Google Scholar] [CrossRef]
- Murdaca, G.; Tonacci, A.; Negrini, S.; Greco, M.; Borro, M.; Puppo, F.; Gangemi, S. Emerging role of vitamin D in autoimmune diseases: An update on evidence and therapeutic implications. Autoimmun. Rev. 2019, 18, 102350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; McCullough, P.A.; Tecson, K.M. Vitamin D deficiency in association with endothelial dysfunction: Implications for patients with COVID-19. Rev. Cardiovasc. Med. 2020, 21, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Hathaway, L.; Mincemoyer, R.; Schenke, W.H.; Kirby, M.; Csako, G.; Waclawiw, M.A.; Panza, J.A.; Cannon, I.R.O. Oral L-arginine in patients with coronary artery disease on medical management. Circulation 2000, 101, 2160–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.M.; Harada, R.; Nair, N.; Balasubramanian, N.; Cooke, J.P. L-arginine supplementation in peripheral artery disease: No benefit and possible harm. Circulation 2003, 116, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalera, F.; Closs, E.I.; Flick, E.; Martens-Lobenhoffer, J.; Boissel, J.P.; Lendeckel, U.; Heimburg, A.; Bode-Böger, S.M. Paradoxical effect of L-arginine: Acceleration of endothelial cell senescence. Biochem. Biophys. Res. Commun. 2009, 386, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Kovamees, O.; Shemyakin, A.; Eriksson, M.; Angelin, B.; Pernow, J. Arginase inhibition improves endothelial function with familial hypercholesterolemia irrespective of their cholesterol level. J. Intern. Med. 2016, 279, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Kovamees, O.; Shemyakin, A.; Checa, A.; Wheelock, C.E.; Lundberg, J.O.; Ostenson, C.-G.; Pernow, J. Arginase inhibition improves microvascular endothelial function in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2016, 101, 3952–3958. [Google Scholar] [CrossRef] [PubMed]
- Madhi, A.; Pernow, J.; Kovamees, O. Arginase inhibition improves endothelial function in age-dependent manner in healthy elderly humans. Rejuvenation Res. 2019, 22, 385–389. [Google Scholar]
- Holowatz, L.A.; Kenney, W.L. Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilation in hypertensive humans. J. Physiol. 2007, 581, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Reid, K.M.; Tsung, A.; Kaizu, T.; Jeyabalan, G.; Ikeda, A.; Shao, L.; Wu, G.; Murase, N.; Geller, D.A. Liver I/R injury is improved by the arginase inhibitor N(omega)-hydroxy-nor-L-arginine (nor-NOHA). Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G512–G527. [Google Scholar] [CrossRef]
- Huynh, H.H.; Harris, E.E.; Chin-Dusting, J.F.P.; Andrews, L.K. The vascular effects of different arginase inhibitors in rat isolated aorta and mesenteric arteries. Br. J. Pharmacol. 2009, 156, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borek, B.; Gajda, T.; Golebiowski, A.; Blaszczyk, R. Boronic acid-based arginase inhibitors in cancer immunotherapy. Bioorg. Med. Chem. 2020, 28, 115658. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, H.; Alsrhani, A.; Zafar, A.; Javed, H.; Junaid, K.; Abdalla, A.E.; Abosalif, K.O.; Ahmed, Z.; Younas, S. COVID-19 and comorbidities: Deleterious impact on infected patients. J. Infect. Public Health 2020, 13, 1833–1839. [Google Scholar] [CrossRef] [PubMed]
- Safaee Fakhr, B.; Wiegand, S.B.; Pinciroli, R.; Gianni, S.; Morais, C.C.A.; Ikeda, T.; Miyazaki, Y.; Marutani, E.; Di Fenza, R.; Larson, G.; et al. High concentrations of nitric oxide inhalation therapy in pregnant patients with severe coronavirus disease 2019 (COVID-19). Obstet. Gynecol. 2020, 136, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Weigand, S.B.; Safaee Fakhr, B.; Carroll, R.W.; Zapol, W.M.; Kacmarek, R.M.; Berra, L. Rescue treatment with high-dose gaseous nitric oxide in spontaneously breathing patients with severe coronavirus disease 2019. Crit. Care Explor. 2020, 2, e0277. [Google Scholar] [CrossRef] [PubMed]
- Safaee Fakhr, B.; Di Fenza, R.; Gianni, S.; Wiegand, S.B.; Miyazaki, Y.; Araujo, C.C.; Gibson, L.E.; Chang, M.G.; Mueller, A.L.; Rodriguez-Lopez, J.M.; et al. Inhaled high dose nitric oxide is a safe and effective respiratory treatment in spontaneous breathing hospitalized patients with COVID-19 pneumonia. Nitric Oxide 2021, 116, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, R.T.; Pollack, C.V., Jr.; Gentile, M.A.; Rahid, M.; Fox, J.C.; Mahaffe, K.W.; Perez, V.D.J. Outpatient inhaled nitric oxide in a patient with vasoreactive idiopathic pulmonary arterial hypertension and COVID-19 infection. Am. J. Respir. Crit. Care Med. 2020, 202, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Abou-Arab, O.; Huette, P.; Debouvries, F.; Dupont, H.; Jounieaux, V.; Mahjoub, Y. Inhaled nitric oxide for critically ill COVID-19 patients: A prospective study. Crit. Care 2020, 24, 645. [Google Scholar] [CrossRef] [PubMed]
- Ziehr, D.R.; Alladina, J.; Wolf, M.E.; Brait, K.L.; Malhotra, A.; La Vita, C.; Berra, L.; Hibbert, K.A.; Hardin, C.C. Respiratory physiology of prone positioning with and without inhaled nitric oxide across the coronavirus disease 2019 acute respiratory distress syndrome severity spectrum. Crit. Care Explor. 2021, 3, e0471. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, G.; Marco, P.; Mongodi, S.; Dammassa, V.; Romito, G.; Mojoli, F. Inhaled nitric oxide in patients admitted to intensive care unit with COVID-19 pneumonia. Crit. Care 2020, 24, 508. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.; Santini, A.; Protti, A.; Andreis, D.T.; Iapichino, G.; Castellani, G.; Rendiniello, V.; Costantini, E.; Cecconi, M. Inhaled nitric oxide in mechanically ventilated patients with COVID-19. J. Crit. Care 2020, 60, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Chandel, A.; Patolia, S.; Ahmad, K.; Aryal, S.; Brown, A.W.; Sahjwani, D.; Khangoora, V.; Shlobin, O.A.; Cameron, P.C.; Singhal, A.; et al. Inhaled nitric oxide via high-flow nasal cannula in patients with acute respiratory failure related to COVID-19. Clin. Med. Insights. Circ. Respir. Pulm. Med. 2021, 15, 1–11. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, Z.; Chen, Y.; Su, H.; Deng, X.; Liu, X.; Fan, Y. Delivery of nitric oxide in the cardiovascular system: Implications for clinical diagnosis and therapy. Int. J. Mol. Sci. 2021, 22, 12166. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Jeong, S.; Ku, S.; Lee, K.; Park, M.H. Use of gasotransmitters for the controlled release of polymer-based nitric oxide carriers in medical applications. J. Control. Release 2018, 279, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug. Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Kapil, V.; Khambata, R.S.; Robertson, A.; Caulfield, M.J.; Ahluwalia, A. Dietary nitrate provides sustained blood pressure lowering in hypertensive patients: A randomized phase 2, double-blind, placebo-controlled study. Hypertension 2015, 65, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karetnikova, E.S.; Jarzebska, N.; Markov, A.G.; Weiss, N.; Lentz, S.R.; Rodionov, R.N. Is homoarginine a protective cardiovascular risk factor? Arter. Thromb. Vasc. Biol. 2019, 39, 869–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atzler, D.; Schonhoff, M.; Cordts, K.; Ortland, I.; Hoppe, J.; Hummel, F.C.; Gerloff, C.; Jaehde, U.; Jagodzinski, A.; Böger, R.H.; et al. Oral supplementation with L-homoarginine in young volunteers. Br. J. Clin. Pharmacol. 2016, 82, 1477–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jud, P.; Gressenberger, P.; Muster, V.; Avian, A.; Meinitzer, A.; Strohmaier, H.; Sourij, H.; Raggam, R.B.; Stradner, M.H.; Demel, U.; et al. Evaluation of endothelial dysfunction and inflammatory vasculopathy after SARS-CoV-2 infection—a cross sectional study. Front. Cardiovasc. Med. 2021, 8, 750887. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.; Michel, L.Y.M.; Balligand, J.-L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durante, W. Targeting Arginine in COVID-19-Induced Immunopathology and Vasculopathy. Metabolites 2022, 12, 240. https://doi.org/10.3390/metabo12030240
Durante W. Targeting Arginine in COVID-19-Induced Immunopathology and Vasculopathy. Metabolites. 2022; 12(3):240. https://doi.org/10.3390/metabo12030240
Chicago/Turabian StyleDurante, William. 2022. "Targeting Arginine in COVID-19-Induced Immunopathology and Vasculopathy" Metabolites 12, no. 3: 240. https://doi.org/10.3390/metabo12030240
APA StyleDurante, W. (2022). Targeting Arginine in COVID-19-Induced Immunopathology and Vasculopathy. Metabolites, 12(3), 240. https://doi.org/10.3390/metabo12030240