
Skeletal Muscle Uncoupling Proteins in Mice Models of Obesity



Abstract
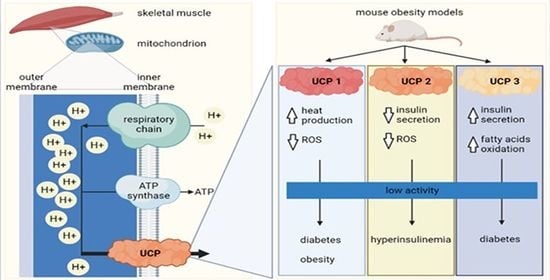
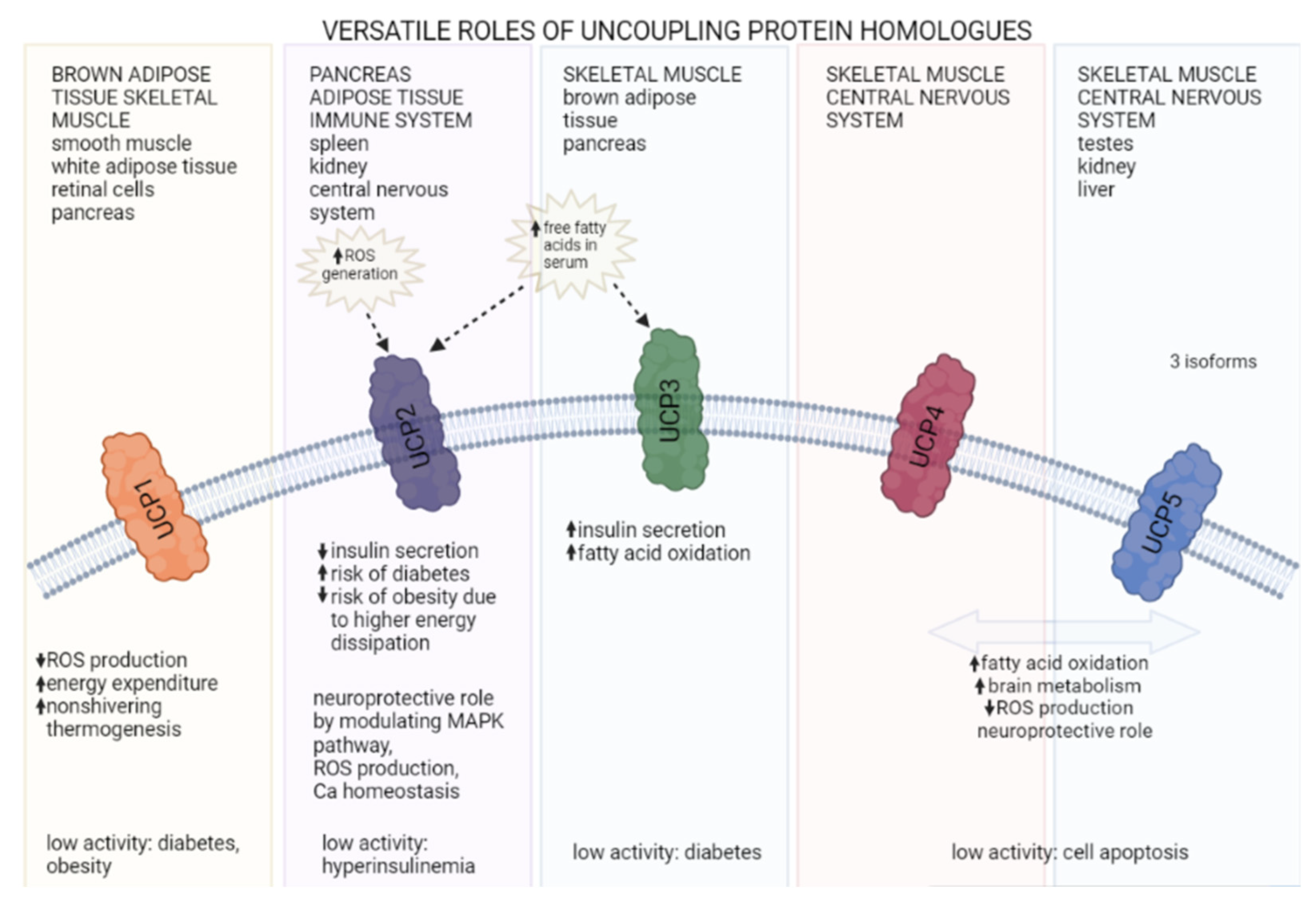
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Metabolic and Physiological Mechanisms of Shivering and Nonshivering Thermogenesis in Skeletal Muscle
2.1. Shivering Thermogenesis
2.2. Nonshivering Thermogenesis
2.2.1. Sarco-Endoplasmic Reticulum Ca2+-ATPase (SERCA) Pump
2.2.2. Myosin ATPase
2.2.3. Creatine Kinase
2.3. Cold Acclimation
3. Coupling, Uncoupling, and Their Interplay in Skeletal Muscle Cells
4. Uncoupling Proteins
4.1. UCP Homologs and Their Roles
4.1.1. UCP1
4.1.2. UCP2
4.1.3. UCP3
4.1.4. Other UCPs
4.2. Sex Differences in UCP Expression
5. UCP2 and Insulin Secretion in Pancreatic Beta-Cells
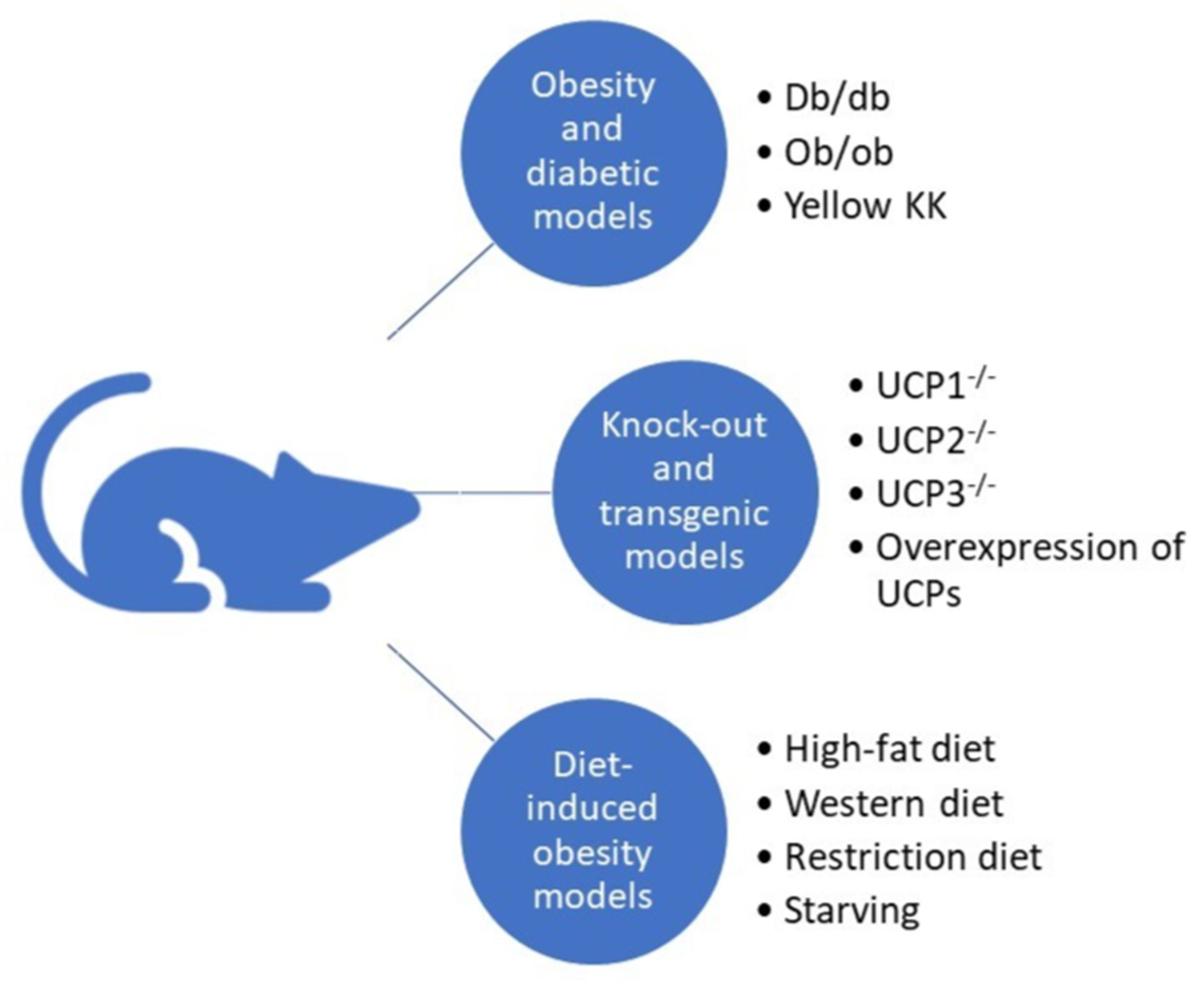
6. UCPs in Mouse Models of Diabetes and Obesity
6.1. Obesity and Diabetic Models
6.2. Transgenic and Knockout Mice
6.3. Diet-Induced Obesity and Diabetic Models
6.4. Translational Precautions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Refinetti, R. The circadian rhythm of body temperature. Front. Biosci. 2010, 15, 564–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talan, I.M.; Ingram, D.K. Age comparisons of body temperature and cold tolerance among different strains of Mus musculus. Mech. Ageing Dev. 1986, 33, 247–256. [Google Scholar] [CrossRef]
- Gonzales, P.; Rikke, B.A. Thermoregulation in mice exhibits genetic variability early in senescence. AGE 2010, 32, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, S.B. Brown adipose tissue and type 2 diabetes. Evol. Med. Public Health 2020, 2020, 70–71. [Google Scholar] [CrossRef] [Green Version]
- Vidal, H.; Langin, D.; Andreelli, F.; Millet, L.; Larrouy, D.; Laville, M. Lack of skeletal muscle uncoupling protein 2 and 3 mRNA induction during fasting in type-2 diabetic subjects. Am. J. Physiol. Content 1999, 277, E830–E837. [Google Scholar] [CrossRef] [PubMed]
- Din, M.U.; Raiko, J.; Saari, T.; Kudomi, N.; Tolvanen, T.; Oikonen, V.; Teuho, J.; Sipilä, H.T.; Savisto, N.; Parkkola, R.; et al. Human brown adipose tissue [15O]O2 PET imaging in the presence and absence of cold stimulus. Eur. J. Pediatr. 2016, 43, 1878–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondin, P.D.; Haman, F. Shivering and nonshivering thermogenesis in skeletal muscles. Handb. Clin. Neurol. 2018, 156, 153–173. [Google Scholar] [PubMed]
- Morrison, S.F. Central neural control of thermoregulation and brown adipose tissue. Auton. Neurosci. 2016, 196, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Yu, H.; Zhao, Z.; Luo, Z.; Chen, J.; Ni, Y.; Jin, R.; Ma, L.; Wang, P.; Zhu, Z.; et al. Activation of the cold-sensing TRPM8 channel triggers UCP1-dependent thermogenesis and prevents obesity. J. Mol. Cell Biol. 2012, 4, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautista, D.M.; Siemens, J.; Glazer, J.M.; Tsuruda, P.R.; Basbaum, A.I.; Stucky, C.L.; Jordt, S.-E.; Julius, D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007, 448, 204–208. [Google Scholar] [CrossRef]
- Romanovsky, A.A. Thermoregulation: Some concepts have changed. Functional architecture of the thermoregulatory system. Am. J. Physiol. Integr. Comp. Physiol. 2007, 292, R37–R46. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Morrison, S.F. Central efferent pathways for cold-defensive and febrile shivering. J. Physiol. 2011, 589, 3641–3658. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.J.; Haman, F. Fuel selection in shivering humans. Acta Physiol. Scand. 2005, 184, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Haman, F. Shivering in the cold: From mechanisms of fuel selection to survival. J. Appl. Physiol. 2006, 100, 1702–1708. [Google Scholar] [CrossRef]
- Haman, F.; Péronnet, F.; Kenny, G.P.; Doucet, E.; Massicotte, D.; Lavoie, C.; Weber, J.-M. Effects of carbohydrate availability on sustained shivering I. Oxidation of plasma glucose, muscle glycogen, and proteins. J. Appl. Physiol. 2004, 96, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Wijers, S.L.J.; Schrauwen, P.; Saris, W.H.M.; Lichtenbelt, W.D.V.M. Human Skeletal Muscle Mitochondrial Uncoupling Is Associated with Cold Induced Adaptive Thermogenesis. PLoS ONE 2008, 3, e1777. [Google Scholar] [CrossRef] [PubMed]
- Bal, N.C.; Maurya, S.K.; Sopariwala, D.H.; Sahoo, S.K.; Gupta, S.C.; Shaikh, S.A.; Pant, M.; Rowland, L.A.; Bombardier, E.; Goonasekera, S.A.; et al. Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nat. Med. 2012, 18, 1575–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamu, D.; Juracic, E.S.; Hall, K.J.; Tupling, A.R. The sarcoplasmic reticulum and SERCA: A nexus for muscular adaptive thermogenesis. Appl. Physiol. Nutr. Metab. 2020, 45, 1–10. [Google Scholar] [CrossRef]
- Shishmarev, D. Excitation-contraction coupling in skeletal muscle: Recent progress and unanswered questions. Biophys. Rev. 2020, 12, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C.; Nakasako, M.; Nomura, H.; Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 2000, 405, 647–655. [Google Scholar] [CrossRef]
- De Meis, L.; Arruda, A.P.; Carvalho, D.P. Role of Sarco/Endoplasmic Reticulum Ca2+-ATPase in Thermogenesis. Biosci. Rep. 2005, 25, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.C.; Bombardier, E.; Vigna, C.; Tupling, A.R. ATP Consumption by Sarcoplasmic Reticulum Ca2+ Pumps Accounts for 40–50% of Resting Metabolic Rate in Mouse Fast and Slow Twitch Skeletal Muscle. PLoS ONE 2013, 8, e68924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytton, J.; Westlin, M.; Burk, S.E.; Shull, G.E.; MacLennan, D.H. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J. Biol. Chem. 1992, 267, 14483–14489. [Google Scholar] [CrossRef]
- De Meis, L. Energy interconversion by the sarcoplasmic reticulum Ca2+-ATPase: ATP hydrolysis, Ca2+ transport, ATP synthesis and heat production. An. Acad. Bras. Ciências 2000, 72, 365–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bal, N.C.; Periasamy, M. Uncoupling of sarcoendoplasmic reticulum calcium ATPase pump activity by sarcolipin as the basis for muscle non-shivering thermogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190135. [Google Scholar] [CrossRef] [Green Version]
- Pant, M.; Bal, N.C.; Periasamy, M. Sarcolipin: A Key Thermogenic and Metabolic Regulator in Skeletal Muscle. Trends Endocrinol. Metab. 2016, 27, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.A.; Franks-Skiba, K.; Chen, S.; Cooke, R. Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc. Natl. Acad. Sci. USA 2009, 107, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Phung, L.A.; Foster, A.D.; Miller, M.S.; Lowe, D.A.; Thomas, D.D. Super-relaxed state of myosin in human skeletal muscle is fiber-type dependent. Am. J. Physiol. Physiol. 2020, 319, C1158–C1162. [Google Scholar] [CrossRef] [PubMed]
- Cooke, R. The role of the myosin ATPase activity in adaptive thermogenesis by skeletal muscle. Biophys. Rev. 2011, 3, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Naber, N.; Cooke, R. The role of the super-relaxed state of myosin in human metabolism. Metab. Open 2021, 9, 100068. [Google Scholar] [CrossRef]
- Wallimann, T.; Wyss, M.; Brdiczka, D.; Nicolay, K.; Eppenberger, H.M. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: The ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 1992, 281, 21–40. [Google Scholar] [CrossRef] [Green Version]
- Kazak, L.; Cohen, P. Creatine metabolism: Energy homeostasis, immunity and cancer biology. Nat. Rev. Endocrinol. 2020, 16, 421–436. [Google Scholar] [CrossRef]
- Berlet, H.; Bonsmann, I.; Birringer, H. Occurrence of free creatine, phosphocreatine and creatine phosphokinase in adipose tissue. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1976, 437, 166–174. [Google Scholar] [CrossRef]
- Kazak, L.; Chouchani, E.T.; Jedrychowski, M.P.; Erickson, B.; Shinoda, K.; Cohen, P.; Vetrivelan, R.; Lu, G.Z.; Laznik-Bogoslavski, D.; Hasenfuss, S.C.; et al. A Creatine-Driven Substrate Cycle Enhances Energy Expenditure and Thermogenesis in Beige Fat. Cell 2015, 163, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Wallimann, T.; Tokarska-Schlattner, M.; Kay, L.; Schlattner, U. Role of creatine and creatine kinase in UCP1-independent adipocyte thermogenesis. Am. J. Physiol. Metab. 2020, 319, E944–E946. [Google Scholar] [CrossRef] [PubMed]
- Rahbani, J.F.; Chouchani, E.T.; Spiegelman, B.M.; Kazak, L. Measurement of Futile Creatine Cycling Using Respirometry. In Brown Adipose Tissue; Humana: New York, NY, USA, 2022; pp. 141–153. [Google Scholar]
- Streijger, F.; Pluk, H.; Oerlemans, F.; Beckers, G.; Bianco, A.C.; Ribeiro, M.O.; Wieringa, B.; Van der Zee, C.E. Mice lacking brain-type creatine kinase activity show defective thermoregulation. Physiol. Behav. 2009, 97, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbani, J.F.; Roesler, A.; Hussain, M.F.; Samborska, B.; Dykstra, C.B.; Tsai, L.; Jedrychowski, M.P.; Vergnes, L.; Reue, K.; Spiegelman, B.M.; et al. Creatine kinase B controls futile creatine cycling in thermogenic fat. Nature 2021, 590, 480–485. [Google Scholar] [CrossRef]
- Bryant, H.J.; Chung, D.J.; Schulte, P.M. Subspecies differences in thermal acclimation of mitochondrial function and the role of uncoupling proteins in killifish. J. Exp. Biol. 2018, 221, jeb.186320. [Google Scholar] [CrossRef] [Green Version]
- Coulson, S.Z.; Robertson, C.E.; Mahalingam, S.; McClelland, G.B. Plasticity of non-shivering thermogenesis and brown adipose tissue in high-altitude deer mice. J. Exp. Biol. 2021, 224, 242279. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of Fatty-Acid-Dependent UCP1 Uncoupling in Brown Fat Mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, S.B.; Leonard, W.R. The evolutionary significance of human brown adipose tissue: Integrating the timescales of adaptation. Evol. Anthr. Issues News Rev. 2021, 1–17. [Google Scholar] [CrossRef]
- Mineo, P.; Cassell, E.; Roberts, M.; Schaeffer, P. Chronic cold acclimation increases thermogenic capacity, non-shivering thermogenesis and muscle citrate synthase activity in both wild-type and brown adipose tissue deficient mice. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2011, 161, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Heldmaier, G.; Klaus, S.; Wiesinger, H.; Friedrichs, U.; Wenzel, M. Cold acclimation and thermogenesis. In Living in the Cold; John Libbey: Momtrouge, France, 1989; pp. 347–358. [Google Scholar]
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Hilse, K.E.; Kalinovich, A.V.; Rupprecht, A.; Smorodchenko, A.; Zeitz, U.; Staniek, K.; Erben, R.G.; Pohl, E.E. The expression of UCP3 directly correlates to UCP1 abundance in brown adipose tissue. Biochim. Biophys. Acta 2016, 1857, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Klingenspor, M.; Ebbinghaus, C.; Hülshorst, G.; Stöhr, S.; Spiegelhalter, F.; Haas, K.; Heldmaier, G. Multiple regulatory steps are involved in the control of lipoprotein lipase activity in brown adipose tissue. J. Lipid Res. 1996, 37, 1685–1695. [Google Scholar] [CrossRef]
- Bukowiecki, L.; Collet, A.J.; Follea, N.; Guay, G.; Jahjah, L. Brown adipose tissue hyperplasia: A fundamental mechanism of adaptation to cold and hyperphagia. Am. J. Physiol. Metab. 1982, 242, E353–E359. [Google Scholar] [CrossRef]
- Yang, X.; Pratley, R.E.; Tokraks, S.; Tataranni, P.A.; Permana, P.A. UCP5/BMCP1 transcript isoforms in human skeletal muscle: Relationship of the short-insert isoform with lipid oxidation and resting metabolic rates. Mol. Genet. Metab. 2002, 75, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.K.; Rennie, D.W.; Park, Y.S. Humans Can Acclimatize to Cold: A Lesson from Korean Women Divers. Physiology 1987, 2, 79–82. [Google Scholar] [CrossRef]
- Schaeffer, P.J.; Villarin, J.J.; Lindstedt, S.L. Chronic Cold Exposure Increases Skeletal Muscle Oxidative Structure and Function in Monodelphis domestica, a Marsupial Lacking Brown Adipose Tissue. Physiol. Biochem. Zool. 2003, 76, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Mitchell, P.; Moyle, J. Chemiosmotic Hypothesis of Oxidative Phosphorylation. Nature 1967, 213, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J. Theor. Biol. 1976, 62, 327–367. [Google Scholar] [CrossRef]
- Mitchell, P. Nobel Lecture, Reimpression de les Prix Nobel en 1978; Nobel Foundation: Stockholm, Sweden, 1979. [Google Scholar]
- Sazanov, L. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Capitanio, G.; Papa, F. The mechanism of coupling between oxido-reduction and proton translocation in respiratory chain enzymes. Biol. Rev. 2017, 93, 322–349. [Google Scholar] [CrossRef] [PubMed]
- von Ballmoos, C.; Wiedenmann, A.; Dimroth, P. Essentials for ATP Synthesis by F1F0 ATP Synthases. Annu. Rev. Biochem. 2009, 78, 649–672. [Google Scholar] [CrossRef] [Green Version]
- Junge, W. Protons, proteins and ATP. In Discoveries in Photosynthesis; Springer: Berlin/Heidelberg, Germany, 2005; pp. 573–595. [Google Scholar]
- Nath, S. Molecular mechanistic insights into uncoupling of ion transport from ATP synthesis. Biophys. Chem. 2018, 242, 15–21. [Google Scholar] [CrossRef]
- Ravera, S.; Panfoli, I.; Aluigi, M.G.; Calzia, D.; Morelli, A. Characterization of myelin sheath F o F 1-ATP synthase and its regulation by IF 1. Cell Biochem. Biophys. 2011, 59, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, M.; Imamura, H.; Nakamura, J.; Yoshida, M. Assessing Actual Contribution of IF1, Inhibitor of Mitochondrial FoF1, to ATP Homeostasis, Cell Growth, Mitochondrial Morphology, and Cell Viability. J. Biol. Chem. 2012, 287, 18781–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, A.M.; Ravera, S.; Calzia, D.; Panfoli, I. An update of the chemiosmotic theory as suggested by possible proton currents inside the coupling membrane. Open Biol. 2019, 9, 180221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolfe, D.F.; Brand, M.D. Contribution of mitochondrial proton leak to skeletal muscle respiration and to standard metabolic rate. Am. J. Physiol. Content 1996, 271, 1380–1389. [Google Scholar] [CrossRef]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolfe, D.F.S.; Brand, M. The Physiological Significance of Mitochondrial Proton Leak in Animal Cells and Tissues. Biosci. Rep. 1997, 17, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ricquier, D. UCP1, the mitochondrial uncoupling protein of brown adipocyte: A personal contribution and a historical perspective. Biochimie 2017, 134, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Bernson, V.S.; Heaton, G.M. The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. In Effectors of Thermogenesis; Springer: Berlin/Heidelberg, Germany, 1978; pp. 89–93. [Google Scholar]
- Ricquier, D.; Bouillaud, F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 2000, 345, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Van Baak, M.A. Meal-induced activation of the sympathetic nervous system and its cardiovascular and thermogenic effects in man. Physiol. Behav. 2008, 94, 178–186. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, S.A.A.; Lichtenbelt, W.V.M.; van Dijk, K.W.; Schrauwen, P. Skeletal muscle mitochondrial uncoupling, adaptive thermogenesis and energy expenditure. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudry, M.J.; Jastroch, M. Molecular evolution of uncoupling proteins and implications for brain function. Neurosci. Lett. 2019, 696, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Géloën, A.; Trayhurn, P. Regulation of the level of uncoupling protein in brown adipose tissue by insulin. Am. J. Physiol. Integr. Comp. Physiol. 1990, 258, R418–R424. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.; Zhang, C.-Y.; Lowell, B.B. The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. Cell Biol. 2005, 6, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, J.; Li, W.-J.; Wang, C.-M. The Role of Uncoupling Proteins in Diabetes Mellitus. J. Diabetes Res. 2013, 2013, 585897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Harper, M.-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, T.C.; Brand, M.D. The reactions catalysed by the mitochondrial uncoupling proteins UCP2 and UCP3. Biochim. Biophys. Acta 2005, 1709, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giralt, M.; Martin, I.; Iglesias, R.; Viñas, O.; Villarroya, F.; Mampel, T. Ontogeny and perinatal modulation of gene expression in rat brown adipose tissue: Unaltered iodothyronine 5′-deiodinase activity is necessary for the response to environmental temperature at birth. Eur. J. Biochem. 1990, 193, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Houštěk, J.; Kopecky, J.; Rychter, Z.; Soukup, T. Uncoupling protein in embryonic brown adipose tissue—Existence of nonthermogenic and thermogenic mitochondria. Biochim. Biophys. Acta 1988, 935, 19–25. [Google Scholar] [CrossRef]
- Shinde, A.B.; Song, A.; Wang, Q.A. Brown Adipose Tissue Heterogeneity, Energy Metabolism, and Beyond. Front. Endocrinol. 2021, 12, 651763. [Google Scholar] [CrossRef]
- Song, A.; Dai, W.; Jang, M.J.; Medrano, L.; Li, Z.; Zhao, H.; Shao, M.; Tan, J.; Li, A.; Ning, T.; et al. Low- and high-thermogenic brown adipocyte subpopulations coexist in murine adipose tissue. J. Clin. Investig. 2019, 130, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Bukowiecki, L.J.; Géloën, A.; Collet, A.J. Proliferation and differentiation of brown adipocytes from interstitial cells during cold acclimation. Am. J. Physiol. Physiol. 1986, 250, C880–C887. [Google Scholar] [CrossRef]
- Lee, Y.; Petkova, A.P.; Konkar, A.A.; Granneman, J.G. Cellular origins of cold-induced brown adipocytes in adult mice. FASEB J. 2015, 29, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Burcelin, R.; Kande, J.; Ricquier, D.; Girard, J. Changes in uncoupling protein and GLUT4 glucose transporter expressions in interscapular brown adipose tissue of diabetic rats: Relative roles of hyperglycaemia and hypoinsulinaemia. Biochem. J. 1993, 291, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Betz, M.J.; Enerbäck, S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat. Rev. Endocrinol. 2018, 14, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Sale, M.M.; Hsu, F.-C.; Palmer, N.D.; Gordon, C.J.; Keene, K.L.; Borgerink, H.M.; Sharma, A.J.; Bergman, R.N.; Taylor, K.D.; Saad, M.F.; et al. The uncoupling protein 1 gene, UCP1, is expressed in mammalian islet cells and associated with acute insulin response to glucose in African American families from the IRAS Family Study. BMC Endocr. Disord. 2007, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adjeitey, C.N.-K.; Mailloux, R.J.; Dekemp, R.A.; Harper, M.-E. Mitochondrial uncoupling in skeletal muscle by UCP1 augments energy expenditure and glutathione content while mitigating ROS production. Am. J. Physiol. Metab. 2013, 305, E405–E415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orava, J.; Nuutila, P.; Lidell, M.E.; Oikonen, V.; Noponen, T.; Viljanen, T.; Scheinin, M.; Taittonen, M.; Niemi, T.; Enerbäck, S.; et al. Different Metabolic Responses of Human Brown Adipose Tissue to Activation by Cold and Insulin. Cell Metab. 2011, 14, 272–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudry, M.J.; Campbell, K.L.; Jastroch, M. Evolution of UCP1. In Brown Adipose Tissue; Springer: Berlin/Heidelberg, Germany, 2018; pp. 127–141. [Google Scholar]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.-J.; Tian, Y.-B.; Cao, Z.-H.; Tao, L.-L.; Zhang, X.; Gao, S.-Z.; Ge, C.-R.; Lin, Q.-Y.; Jois, M. The polymorphisms of UCP1 genes associated with fat metabolism, obesity and diabetes. Mol. Biol. Rep. 2010, 37, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Hamann, A.; Tafel, J.; Büsing, B.; Münzberg, H.; Hinney, A.; Mayer, H.; Siegfried, W.; Ricquier, D.; Greten, H.; Hebebrand, J.; et al. Analysis of the uncoupling protein-1 (UCP1) gene in obese and lean subjects: Identification of four amino acid variants. Int. J. Obes. 1998, 22, 939–941. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Cho, D.-Y.; Kim, Y.J.; Choi, S.M.; Kim, J.Y.; Shin, S.U.; Yoon, Y.S. The finding of new genetic polymorphism of UCP-1 A-1766G and its effects on body fat accumulation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2005, 1741, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Brun, S.; Carmona, M.C.; Mampel, T.; Viñas, O.; Giralt, M.; Iglesias, R.; Villarroya, F. Uncoupling protein-3 gene expression in skeletal muscle during development is regulated by nutritional factors that alter circulating non-esterified fatty acids. FEBS Lett. 1999, 453, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-Y.; Baffy, G.; Perret, P.; Krauss, S.; Peroni, O.; Grujic, D.; Hagen, T.; Vidal-Puig, A.J.; Boss, O.; Kim, Y.-B.; et al. Uncoupling Protein-2 Negatively Regulates Insulin Secretion and Is a Major Link between Obesity, β Cell Dysfunction, and Type 2 Diabetes. Cell 2001, 105, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Krauss, S.; Zhang, C.Y.; Scorrano, L.; Dalgaard, L.T.; St-Pierre, J.; Grey, S.T.; Lowell, B.B. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic β cell dysfunction. J. Clin. Investig. 2003, 112, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.A.; Zhao, Z.; Turk, J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp. Diabetes Res. 2012, 2012, 703538. [Google Scholar] [CrossRef] [Green Version]
- De Souza, C.T.; Araújo, E.P.; Stoppiglia, L.F.; Pauli, J.R.; Ropelle, E.; Rocco, S.A.; Marin, R.M.; Franchini, K.G.; Carvalheira, J.B.; Saad, M.J.; et al. Inhibition of UCP2 expression reverses diet-induced diabetes mellitus by effects on both insulin secretion and action. FASEB J. 2007, 21, 1153–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson-Doucette, C.A.; Sultan, S.; Allister, E.M.; Wikstrom, J.D.; Koshkin, V.; Bhattacharjee, A.; Prentice, K.J.; Sereda, S.B.; Shirihai, O.S.; Wheeler, M.B. β-Cell Uncoupling Protein 2 Regulates Reactive Oxygen Species Production, Which Influences Both Insulin and Glucagon Secretion. Diabetes 2011, 60, 2710–2719. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Cai, T.; Zhou, Y.; Wang, Y.; Wang, H.; Shen, Z.; Xia, W.; Liu, X.; Ding, B.; Luo, Y.; et al. Ethnicity Differences in the Association of UCP1-3826A/G, UCP2-866G/A and Ala55Val, and UCP3-55C/T Polymorphisms with Type 2 Diabetes Mellitus Susceptibility: An Updated Meta-Analysis. BioMed Res. Int. 2021, 2021, 3482879. [Google Scholar] [CrossRef]
- Emre, Y.; Nübel, T. Uncoupling protein UCP2: When mitochondrial activity meets immunity. FEBS Lett. 2010, 584, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Diano, S.; Horvath, T.L. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol. Med. 2012, 18, 52–58. [Google Scholar] [CrossRef]
- Mehta, S.L.; Li, P.A. Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. J. Cereb. Blood Flow Metab. 2009, 29, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Boss, O.; Samec, S.; Paoloni-Giacobino, A.; Rossier, C.; Dulloo, A.; Seydoux, J.; Muzzin, P.; Giacobino, J.-P. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997, 408, 39–42. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Puig, A.; Solanes, G.; Grujic, D.; Flier, J.S.; Lowell, B.B. UCP3: An Uncoupling Protein Homologue Expressed Preferentially and Abundantly in Skeletal Muscle and Brown Adipose Tissue. Biochem. Biophys. Res. Commun. 1997, 235, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.-W.; He, Y.; Karas, M.; Reitman, M. Uncoupling Protein-3 Is a Mediator of Thermogenesis Regulated by Thyroid Hormone, β3-Adrenergic Agonists, and Leptin. J. Biol. Chem. 1997, 272, 24129–24132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal-Puig, A.; Grujic, D.; Zhang, C.-Y.; Hagen, T.; Boss, O.; Ido, Y.; Szczepanik, A.; Wade, J.; Mootha, V.; Cortright, R.; et al. Energy Metabolism in Uncoupling Protein 3 Gene Knockout Mice. J. Biol. Chem. 2000, 275, 16258–16266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzu, V.; Jastroch, M.; Divakaruni, A.S.; Brand, M.D. The regulation and turnover of mitochondrial uncoupling proteins. Biochim. Biophys. Acta 2010, 1797, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Alán, L.; Smolková, K.; Kronusová, E.; Šantorová, J.; Ježek, P. Absolute levels of transcripts for mitochondrial uncoupling proteins UCP2, UCP3, UCP4, and UCP5 show different patterns in rat and mice tissues. J. Bioenerg. Biomembr. 2009, 41, 71–78. [Google Scholar] [CrossRef]
- Weigle, D.S.; Selfridge, L.E.; Schwartz, M.W.; Seeley, R.J.; Cummings, D.E.; Havel, P.J.; Kuijper, J.L.; BeltrandelRio, H. Elevated free fatty acids induce uncoupling protein 3 expression in muscle: A potential explanation for the effect of fasting. Diabetes 1998, 47, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedraza, N.; Solanes, G.; Carmona, M.C.; Iglesias, R.; Viñas, O.; Mampel, T.; Vazquez, M.; Giralt, M.; Villarroya, F. Impaired expression of the uncoupling protein-3 gene in skeletal muscle during lactation: Fibrates and troglitazone reverse lactation-induced downregulation of the uncoupling protein-3 gene. Diabetes 2000, 49, 1224–1230. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Maedler, K.; Shu, L.; Haataja, L. UCP-2 and UCP-3 Proteins Are Differentially Regulated in Pancreatic Beta-Cells. PLoS ONE 2008, 3, e1397. [Google Scholar] [CrossRef]
- Costford, S.R.; Chaudhry, S.N.; Crawford, S.A.; Salkhordeh, M.; Harper, M.-E. Long-term high-fat feeding induces greater fat storage in mice lacking UCP3. Am. J. Physiol. Metab. 2008, 295, E1018–E1024. [Google Scholar] [CrossRef]
- Holloway, G.P.; Jain, S.S.; Bezaire, V.; Han, X.X.; Glatz, J.F.C.; Luiken, J.J.F.P.; Harper, M.-E.; Bonen, A. FAT/CD36-null mice reveal that mitochondrial FAT/CD36 is required to upregulate mitochondrial fatty acid oxidation in contracting muscle. Am. J. Physiol. Integr. Comp. Physiol. 2009, 297, R960–R967. [Google Scholar] [CrossRef]
- Schrauwen, P.; Mensink, M.; Schaart, G.; Moonen-Kornips, E.; Sels, J.-P.; Blaak, E.E.; Russell, A.P.; Hesselink, M.K.C. Reduced Skeletal Muscle Uncoupling Protein-3 Content in Prediabetic Subjects and Type 2 Diabetic Patients: Restoration by Rosiglitazone Treatment. J. Clin. Endocrinol. Metab. 2006, 91, 1520–1525. [Google Scholar] [CrossRef] [Green Version]
- Nabben, M.; Hoeks, J. Mitochondrial uncoupling protein 3 and its role in cardiac- and skeletal muscle metabolism. Physiol. Behav. 2008, 94, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Azzu, V.; Brand, M.D. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem. Sci. 2010, 35, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, S.; Mozo, J.; Dujardin, G.; Emre, Y.; Masscheleyn, S.; Ricquier, D.; Cassard-Doulcier, A.-M. UCP2 is a mitochondrial transporter with an unusual very short half-life. FEBS Lett. 2007, 581, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Herron, D.; Gianotti, M.; Palou, A.; Cannon, B.; Nedergaard, J. Induction and degradation of the uncoupling protein thermogenin in brown adipocytes in vitro and in vivo. Evidence for a rapidly degradable pool. Biochem. J. 1992, 284, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Pecqueur, C.; Alves-Guerra, M.-C.; Gelly, C.; Lévi-Meyrueis, C.; Couplan, E.; Collins, S.; Ricquier, D.; Bouillaud, F.; Miroux, B. Uncoupling Protein 2, in Vivo Distribution, Induction upon Oxidative Stress, and Evidence for Translational Regulation. J. Biol. Chem. 2001, 276, 8705–8712. [Google Scholar] [CrossRef] [Green Version]
- Sanchis, D.; Fleury, C.; Chomiki, N.; Goubern, M.; Huang, Q.; Neverova, M.; Grégoire, F.; Easlick, J.; Raimbault, S.; Lévi-Meyrueis, C.; et al. BMCP1, a Novel Mitochondrial Carrier with High Expression in the Central Nervous System of Humans and Rodents, and Respiration Uncoupling Activity in Recombinant Yeast. J. Biol. Chem. 1998, 273, 34611–34615. [Google Scholar] [CrossRef] [Green Version]
- Mao, W.; Yu, X.X.; Zhong, A.; Li, W.; Brush, J.; Sherwood, S.W.; Adams, S.H.; Pan, G. UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett. 1999, 443, 326–330. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, D.B.; Ho, P.W.; Ho, J.W.; Liu, H.; So, D.H.; Tse, H.; Chan, K.; Ho, S. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): Structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2012, 2, 468–478. [Google Scholar] [CrossRef]
- Rodríguez, A.M.; Palou, A. Uncoupling proteins: Gender dependence and their relation to body weight control. Int. J. Obes. 2004, 28, 500–502. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M.; Valencia, T.; Brun-Zinkernagel, A.-M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor β. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef] [Green Version]
- Eason, J.M.; Schwartz, G.A.; Pavlath, G.K.; English, A.W. Sexually dimorphic expression of myosin heavy chains in the adult mouse masseter. J. Appl. Physiol. 2000, 89, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.M.; Monjo, M.; Roca, P.; Palou, A. Opposite actions of testosterone and progesterone on UCP1 mRNA expression in cultured brown adipocytes. Cell. Mol. Life Sci. CMLS 2002, 59, 1714–1723. [Google Scholar] [CrossRef]
- Valle, A.; García-Palmer, F.; Oliver, J.; Roca, P. Sex Differences in Brown Adipose Tissue Thermogenic Features During Caloric Restriction. Cell. Physiol. Biochem. 2007, 19, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Moschinger, M.; Hilse, K.E.; Rupprecht, A.; Zeitz, U.; Erben, R.G.; Rülicke, T.; Pohl, E.E. Age-related sex differences in the expression of important disease-linked mitochondrial proteins in mice. Biol. Sex Differ. 2019, 10, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguer, C.; Fiehn, O.; Seifert, E.L.; Bézaire, V.; Meissen, J.K.; Daniels, A.; Scott, K.; Renaud, J.; Padilla, M.; Bickel, D.R.; et al. Muscle uncoupling protein 3 overexpression mimics endurance training and reduces circulating biomarkers of incomplete β-oxidation. FASEB J. 2013, 27, 4213–4225. [Google Scholar] [CrossRef] [Green Version]
- Bazhan, N.; Jakovleva, T.; Feofanova, N.; Denisova, E.; Dubinina, A.; Sitnikova, N.; Makarova, E. Sex Differences in Liver, Adipose Tissue, and Muscle Transcriptional Response to Fasting and Refeeding in Mice. Cells 2019, 8, 1529. [Google Scholar] [CrossRef] [Green Version]
- Erecińska, M.; Bryła, J.; Michalik, M.; Meglasson, M.D.; Nelson, D. Energy metabolism in islets of Langerhans. Biochim. Biophys. Acta (BBA)-Bioenerg. 1992, 1101, 273–295. [Google Scholar] [CrossRef]
- Henquin, J.-C. The dual control of insulin secretion by glucose involves triggering and amplifying pathways in β-cells. Diabetes Res. Clin. Pract. 2011, 93, S27–S31. [Google Scholar] [CrossRef]
- Klemen, M.S.; Dolenšek, J.; Rupnik, M.S.; Stožer, A. The triggering pathway to insulin secretion: Functional similarities and differences between the human and the mouse β cells and their translational relevance. Islets 2017, 9, 109–139. [Google Scholar] [CrossRef] [Green Version]
- Stožer, A.; Leitgeb, E.P.; Pohorec, V.; Dolenšek, J.; Bombek, L.K.; Gosak, M.; Klemen, M.S. The Role of cAMP in Beta Cell Stimulus-Secretion and Inter-cellular Coupling. Cells 2021, 10, 1658. [Google Scholar] [CrossRef]
- Broche, B.; Ben Fradj, S.; Aguilar, E.; Sancerni, T.; Bénard, M.; Makaci, F.; Berthault, C.; Scharfmann, R.; Alves-Guerra, M.-C.; Duvillié, B. Mitochondrial Protein UCP2 Controls Pancreas Development. Diabetes 2017, 67, 78–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, M.; Schilf, P.; Lange, F.; Mayer, J.; Reichart, G.; Maity, P.; Jöhren, O.; Schwaninger, M.; Scharffetter-Kochanek, K.; Sina, C.; et al. Uncoupling protein 2 protects mice from aging. Mitochondrion 2016, 30, 42–50. [Google Scholar] [CrossRef]
- Chan, C.B.; MacDonald, P.E.; Saleh, M.C.; Johns, D.C.; Marbàn, E.; Wheeler, M.B. Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin secretion from rat islets. Diabetes 1999, 48, 1482–1486. [Google Scholar] [CrossRef]
- Krauss, S.; Zhang, C.-Y.; Lowell, B.B. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc. Natl. Acad. Sci. USA 2001, 99, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lameloise, N.; Muzzin, P.; Prentki, M.; Assimacopoulos-Jeannet, F. Uncoupling Protein 2: A Possible Link Between Fatty Acid Excess and Impaired Glucose-Induced Insulin Secretion? Diabetes 2001, 50, 803–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.B.; de Leo, D.; Joseph, J.W.; McQuaid, T.S.; Ha, X.F.; Xu, F.; Tsushima, R.G.; Pennefather, P.S.; Salapatek, A.M.F.; Wheeler, M.B. Increased uncoupling protein-2 levels in β-cells are associated with impaired glucose-stimulated insulin secretion: Mechanism of action. Diabetes 2001, 50, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.B.; Saleh, M.C.; Koshkin, V.; Wheeler, M.B. Uncoupling Protein 2 and Islet Function. Diabetes 2004, 53 (Suppl. 1), S136–S142. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.W.; Koshkin, V.; Zhang, C.-Y.; Wang, J.; Lowell, B.B.; Chan, C.B.; Wheeler, M.B. Uncoupling Protein 2 Knockout Mice Have Enhanced Insulin Secretory Capacity After a High-Fat Diet. Diabetes 2002, 51, 3211–3219. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.W.; Koshkin, V.; Saleh, M.C.; Sivitz, W.I.; Zhang, C.-Y.; Lowell, B.B.; Chan, C.B.; Wheeler, M.B. Free Fatty Acid-induced β-Cell Defects Are Dependent on Uncoupling Protein 2 Expression. J. Biol. Chem. 2004, 279, 51049–51056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.C.; Wheeler, M.B.; Chan, C.B. Endogenous islet uncoupling protein-2 expression and loss of glucose homeostasis in ob/ob mice. J. Endocrinol. 2006, 190, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Yoshimatsu, H.; Chiba, S.; Sakata, T. Impaired response of UCP family to cold exposure in diabetic (db/db) mice. Am. J. Physiol. Integr. Comp. Physiol. 2000, 279, R1305–R1309. [Google Scholar] [CrossRef] [PubMed]
- Enerbäck, S.; Jacobsson, A.; Simpson, E.; Guerra, C.; Yamashita, H.; Harper, M.-E.; Kozak, L.P. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nat. 1997, 387, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; More, K.J.; Breitbart, R.E.; et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in db/db Mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Bai, C.; Chen, F.; Wang, R.; MacDonald, T.; Gu, M.; Zhang, Q.; Morsy, M.A.; Caskey, C. Uncoupling protein-3: A muscle-specific gene upregulated by leptin in ob/ob mice. Gene 1998, 207, 1–7. [Google Scholar] [CrossRef]
- Scarpace, P.J.; Matheny, M. Leptin induction of UCP1 gene expression is dependent on sympathetic innervation. Am. J. Physiol. Endocrinol. Metab. 1998, 275, E259–E264. [Google Scholar] [CrossRef]
- Samec, S.; Seydoux, J.; Dulloo, A.G. Post-starvation gene expression of skeletal muscle uncoupling protein 2 and uncoupling protein 3 in response to dietary fat levels and fatty acid composition: A link with insulin resistance. Diabetes 1999, 48, 436–441. [Google Scholar] [CrossRef]
- Moore, G.B.; Himms-Hagen, J.; Harper, M.-E.; Clapham, J.C. Overexpression of UCP-3 in Skeletal Muscle of Mice Results in Increased Expression of Mitochondrial Thioesterase mRNA. Biochem. Biophys. Res. Commun. 2001, 283, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Lafontan, M.; Berlan, M. Fat cell adrenergic receptors and the control of white and brown fat cell function. J. Lipid Res. 1993, 34, 1057–1091. [Google Scholar] [CrossRef]
- Yoshida, T.; Sakane, N.; Wakabayashi, Y.; Umekawa, T.; Kondo, M. Anti-obesity and anti-diabetic effects of CL 316, 243, a highly specific β3-adrenoceptor agonist, in yellow KK mice. Life Sci. 1994, 54, 491–498. [Google Scholar] [CrossRef]
- Nagase, I.; Yoshida, T.; Kumamoto, K.; Umekawa, T.; Sakane, N.; Nikami, H.; Kawada, T.; Saito, M. Expression of uncoupling protein in skeletal muscle and white fat of obese mice treated with thermogenic beta 3-adrenergic agonist. J. Clin. Investig. 1996, 97, 2898–2904. [Google Scholar] [CrossRef] [Green Version]
- Argyropoulos, G.; Harper, M.-E. Invited Review: Uncoupling proteins and thermoregulation. J. Appl. Physiol. 2002, 92, 2187–2198. [Google Scholar] [CrossRef] [Green Version]
- Monemdjou, S.; Kozak, L.P.; Harper, M.-E. Mitochondrial proton leak in brown adipose tissue mitochondria of Ucp1-deficient mice is GDP insensitive. Am. J. Physiol. Content 1999, 276, E1073–E1082. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Nolte, L.A.; Ju, J.-S.; Han, D.H.; Coleman, T.; Holloszy, J.O.; Semenkovich, C.F. Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat. Med. 2000, 6, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Gates, A.C.; Bernal-Mizrachi, C.; Chinault, S.L.; Feng, C.; Schneider, J.G.; Coleman, T.; Malone, J.P.; Townsend, R.R.; Chakravarthy, M.V.; Semenkovich, C.F. Respiratory Uncoupling in Skeletal Muscle Delays Death and Diminishes Age-Related Disease. Cell Metab. 2007, 6, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giralt, M.; Villarroya, F. Mitochondrial uncoupling and the regulation of glucose homeostasis. Curr. Diabetes Rev. 2017, 13, 386–394. [Google Scholar] [CrossRef]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.-C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Horvath, T.L.; Diano, S.; Miyamoto, S.; Barry, S.; Gatti, S.; Alberati, D.; Livak, F.; Lombardi, A.; Moreno, M.; Goglia, F.; et al. Uncoupling proteins-2 and 3 influence obesity and inflammation in transgenic mice. Int. J. Obes. 2003, 27, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, D.-W.; Monemdjou, S.; Gavrilova, O.; Leon, L.R.; Marcus-Samuels, B.; Chou, C.J.; Everett, C.; Kozak, L.P.; Li, C.; Deng, C.; et al. Lack of Obesity and Normal Response to Fasting and Thyroid Hormone in Mice Lacking Uncoupling Protein-3. J. Biol. Chem. 2000, 275, 16251–16257. [Google Scholar] [CrossRef] [Green Version]
- Clapham, J.C.; Arch, J.R.S.; Chapman, H.; Haynes, A.; Lister, C.; Moore, G.B.T.; Piercy, V.; Carter, S.A.; Lehner, I.; Smith, S.A.; et al. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature 2000, 406, 415–418. [Google Scholar] [CrossRef]
- Clapham, J.C.; Coulthard, V.H.; Moore, G.B. Concordant mRNA Expression of UCP-3, but Not UCP-2, with Mitochondrial Thioesterase-1 in Brown Adipose Tissue and Skeletal Muscle in db/db Diabetic Mice. Biochem. Biophys. Res. Commun. 2001, 287, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Bezaire, V.; Spriet, L.L.; Campbell, S.; Sabet, N.; Gerrits, M.; Bonen, A.; Harper, M. Constitutive UCP3 overexpression at physiological levels increases mouse skeletal muscle capacity for fatty acid transport and oxidation. FASEB J. 2005, 19, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Winn, N.; Vieira-Potter, V.J.; Gastecki, M.L.; Welly, R.J.; Scroggins, R.J.; Zidon, T.M.; Gaines, T.L.; Woodford, M.L.; Karasseva, N.G.; Kanaley, J.A.; et al. Loss of UCP1 exacerbates Western diet-induced glycemic dysregulation independent of changes in body weight in female mice. Am. J. Physiol. Integr. Comp. Physiol. 2017, 312, R74–R84. [Google Scholar] [CrossRef] [PubMed]
- Warden, C.H.; Fisler, J.S.; Shoemaker, S.M.; Wen, P.Z.; Svenson, K.L.; Pace, M.J.; Lusis, A.J. Identification of four chromosomal loci determining obesity in a multifactorial mouse model. J. Clin. Investig. 1995, 95, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Okabe, M.; Natsume, M.; Ashida, H. Prevention mechanisms of glucose intolerance and obesity by cacao liquor procyanidin extract in high-fat diet-fed C57BL/6 mice. Arch. Biochem. Biophys. 2012, 527, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Wang, S.; Petro, A.E.; Sanchis, D.; Raimbault, S.; Ricquier, D.; Collins, S. Diet-induced changes in uncoupling proteins in obesity-prone and obesity-resistant strains of mice. Proc. Natl. Acad. Sci. USA 1998, 95, 4061–4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bézaire, V.; Hofmann, W.; Kramer, J.K.G.; Kozak, L.P.; Harper, M.-E. Effects of fasting on muscle mitochondrial energetics and fatty acid metabolism in Ucp3(−/−) and wild-type mice. Am. J. Physiol. Metab. 2001, 281, E975–E982. [Google Scholar] [CrossRef] [Green Version]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef]
- Aquilano, K.; Sciarretta, F.; Turchi, R.; Li, B.-H.; Rosina, M.; Ceci, V.; Guidobaldi, G.; Arena, S.; D’Ambrosio, C.; Audano, M.; et al. Low-protein/high-carbohydrate diet induces AMPK-dependent canonical and non-canonical thermogenesis in subcutaneous adipose tissue. Redox Biol. 2020, 36, 101633. [Google Scholar] [CrossRef]
- Cerletti, M.; Jang, Y.C.; Finley, L.W.; Haigis, M.C.; Wagers, A.J. Short-Term Calorie Restriction Enhances Skeletal Muscle Stem Cell Function. Cell Stem Cell 2012, 10, 515–519. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reggio, A.; Rosina, M.; Krahmer, N.; Palma, A.; Petrilli, L.L.; Maiolatesi, G.; Massacci, G.; Salvatori, I.; Valle, C.; Testa, S.; et al. Metabolic reprogramming of fibro/adipogenic progenitors facilitates muscle regeneration. Life Sci. Alliance 2020, 3, e202000646. [Google Scholar] [CrossRef]
- Lee, C.; Liu, M.; Agha, O.; Kim, H.T.; Liu, X.; Feeley, B.T. Beige fibro-adipogenic progenitor transplantation reduces muscle degeneration and improves function in a mouse model of delayed repair of rotator cuff tears. J. Shoulder Elb. Surg. 2020, 29, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, Z.; Lee, C.; Nguyen, C.; Lee, L.; Kim, H.; Feeley, B.; Liu, X. Brown/Beige Fat Activation after Skeletal Muscle Ischemia-Reperfusion Injury. Muscles Ligaments Tendons J. (MLTJ) 2020, 10, 579–588. [Google Scholar] [CrossRef]
- Bryniarski, A.R.; Meyer, G.A. Brown Fat Promotes Muscle Growth During Regeneration. J. Orthop. Res. 2019, 37, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Shabrova, E.V.; Tarnopolsky, O.; Singh, A.P.; Plutzky, J.; Vorsa, N.; Quadro, L. Insights into the Molecular Mechanisms of the Anti-Atherogenic Actions of Flavonoids in Normal and Obese Mice. PLoS ONE 2011, 6, e24634. [Google Scholar] [CrossRef] [Green Version]
- Minakawa, M.; Miura, Y.; Yagasaki, K. Piceatannol, a resveratrol derivative, promotes glucose uptake through glucose transporter 4 translocation to plasma membrane in L6 myocytes and suppresses blood glucose levels in type 2 diabetic model db/db mice. Biochem. Biophys. Res. Commun. 2012, 422, 469–475. [Google Scholar] [CrossRef]
- Putman, C.T.; Kiricsi, M.; Pearcey, J.; MacLean, I.M.; Bamford, J.A.; Murdoch, G.K.; Dixon, W.T.; Pette, D. AMPK activation increases uncoupling protein-3 expression and mitochondrial enzyme activities in rat muscle without fibre type transitions. J. Physiol. 2003, 551, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Do, G.-M.; Jung, U.J.; Park, H.-J.; Kwon, E.-Y.; Jeon, S.-M.; McGregor, R.A.; Choi, M.-S. Resveratrol ameliorates diabetes-related metabolic changes via activation of AMP-activated protein kinase and its downstream targets in db/db mice. Mol. Nutr. Food Res. 2012, 56, 1282–1291. [Google Scholar] [CrossRef]
- Tomaru, M.; Takano, H.; Osakabe, N.; Yasuda, A.; Inoue, K.-I.; Yanagisawa, R.; Ohwatari, T.; Uematsu, H. Dietary supplementation with cacao liquor proanthocyanidins prevents elevation of blood glucose levels in diabetic obese mice. Nutrition 2007, 23, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-L.; Liu, L. Effect of metformin on stem cells: Molecular mechanism and clinical prospect. World J. Stem Cells 2020, 12, 1455–1473. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Jasmin, B.J. Metformin increases peroxisome proliferator–activated receptor γ Co-activator-1α and utrophin a expression in dystrophic skeletal muscle. Muscle Nerve 2015, 52, 139–142. [Google Scholar] [CrossRef]
- Langone, F.; Cannata, S.; Fuoco, C.; Barbato, D.L.; Testa, S.; Nardozza, A.P.; Ciriolo, M.R.; Castagnoli, L.; Gargioli, C.; Cesareni, G. Metformin Protects Skeletal Muscle from Cardiotoxin Induced Degeneration. PLoS ONE 2014, 9, e114018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlidou, T.; Marinkovic, M.; Rosina, M.; Fuoco, C.; Vumbaca, S.; Gargioli, C.; Castagnoli, L.; Cesareni, G. Metformin Delays Satellite Cell Activation and Maintains Quiescence. Stem Cells Int. 2019, 2019, 5980465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitman, M.L. Of mice and men—Environmental temperature, body temperature, and treatment of obesity. FEBS Lett. 2018, 592, 2098–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C. Thermal physiology of laboratory mice: Defining thermoneutrality. J. Therm. Biol. 2012, 37, 654–685. [Google Scholar] [CrossRef]
- Abreu-Vieira, G.; Xiao, C.; Gavrilova, O.; Reitman, M.L. Integration of body temperature into the analysis of energy expenditure in the mouse. Mol. Metab. 2015, 4, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Brychta, R.J.; Chen, K.Y. Cold-induced thermogenesis in humans. Eur. J. Clin. Nutr. 2016, 71, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Karp, C. Unstressing intemperate models: How cold stress undermines mouse modeling. J. Exp. Med. 2012, 209, 1069–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Cannon, B.; Nedergaard, J. Optimal housing temperatures for mice to mimic the thermal environment of humans: An experimental study. Mol. Metab. 2018, 7, 161–170. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Križančić Bombek, L.; Čater, M. Skeletal Muscle Uncoupling Proteins in Mice Models of Obesity. Metabolites 2022, 12, 259. https://doi.org/10.3390/metabo12030259
Križančić Bombek L, Čater M. Skeletal Muscle Uncoupling Proteins in Mice Models of Obesity. Metabolites. 2022; 12(3):259. https://doi.org/10.3390/metabo12030259
Chicago/Turabian StyleKrižančić Bombek, Lidija, and Maša Čater. 2022. "Skeletal Muscle Uncoupling Proteins in Mice Models of Obesity" Metabolites 12, no. 3: 259. https://doi.org/10.3390/metabo12030259
APA StyleKrižančić Bombek, L., & Čater, M. (2022). Skeletal Muscle Uncoupling Proteins in Mice Models of Obesity. Metabolites, 12(3), 259. https://doi.org/10.3390/metabo12030259