
Effects of Anticancer Agent P-bi-TAT on Gene Expression Link the Integrin Thyroid Hormone Receptor to Expression of Stemness and Energy Metabolism Genes in Cancer Cells

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Mechanisms of Anticancer Activities of P-bi-TAT Revealed by Genome-Wide Expression Profiling of a Primary Culture of Human GBM Cells (PC-GBM)
2.2. Examples of Specific GBM Driver Genes Important to Regulation of Cell Division, Energy Metabolism and Signal Transductions of Cancer Survival Pathways Whose Expression Is Affected by P-bi-TAT
2.3. P-bi-TAT Markedly Affects the Transcriptional Architecture of the Energy-Metabolism-Sustaining Life-Support Infrastructure of Cancer Cells
2.4. Expression of ATP Synthase Genes Is Altered by Exposure of GBM Cells to P-bi-TAT
2.5. Expression of NADH Dehydrogenase Genes in Response to Exposure of GBM Cells to P-bi-TAT
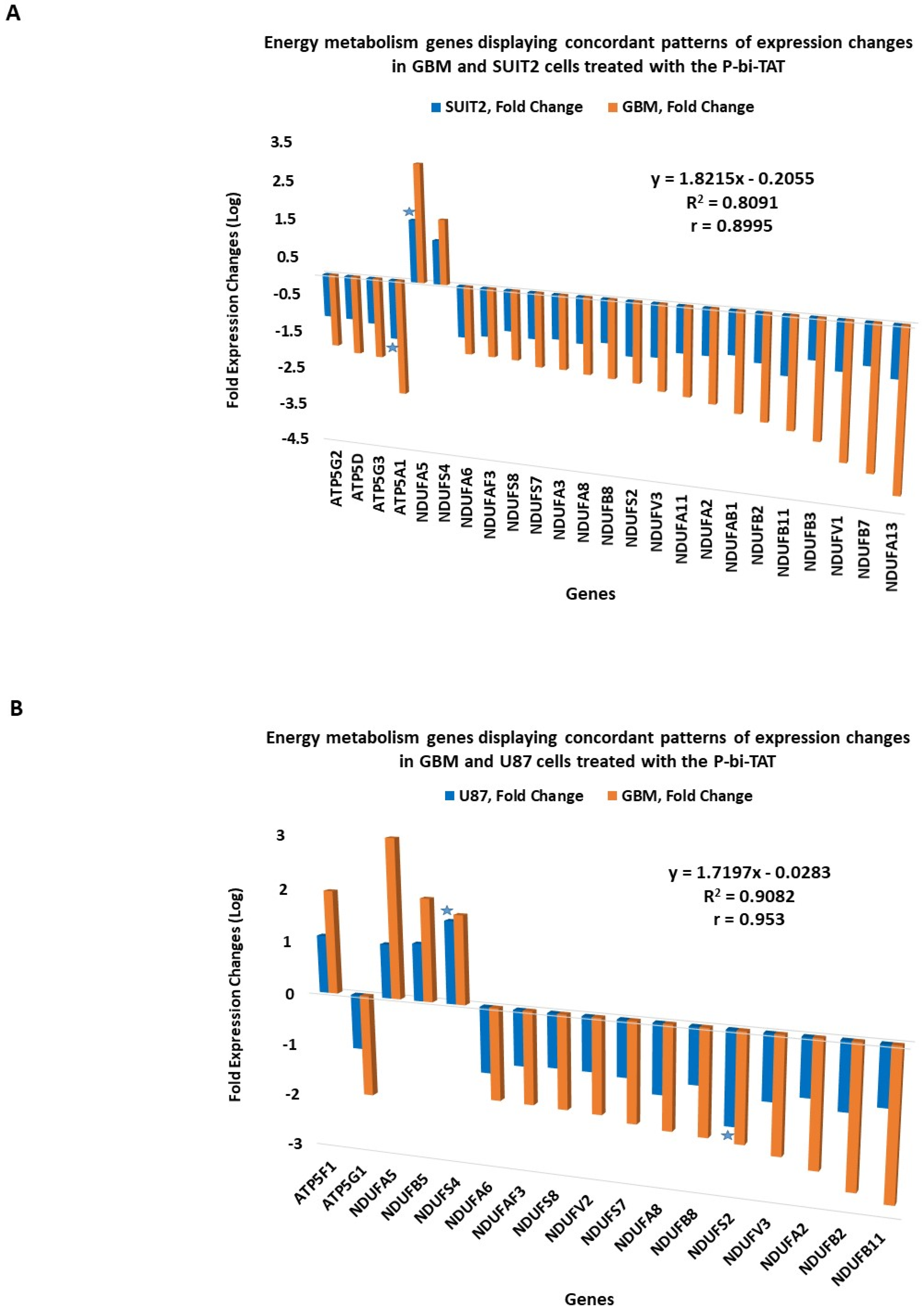
2.6. Concordant Patterns of Expression Changes of Energy Metabolism Genes in Human Cancer Cell Lines in Response to P-bi-TAT
2.7. Concordance of Biological Activities and Molecular Mechanisms of Actions of P-bi-TAT
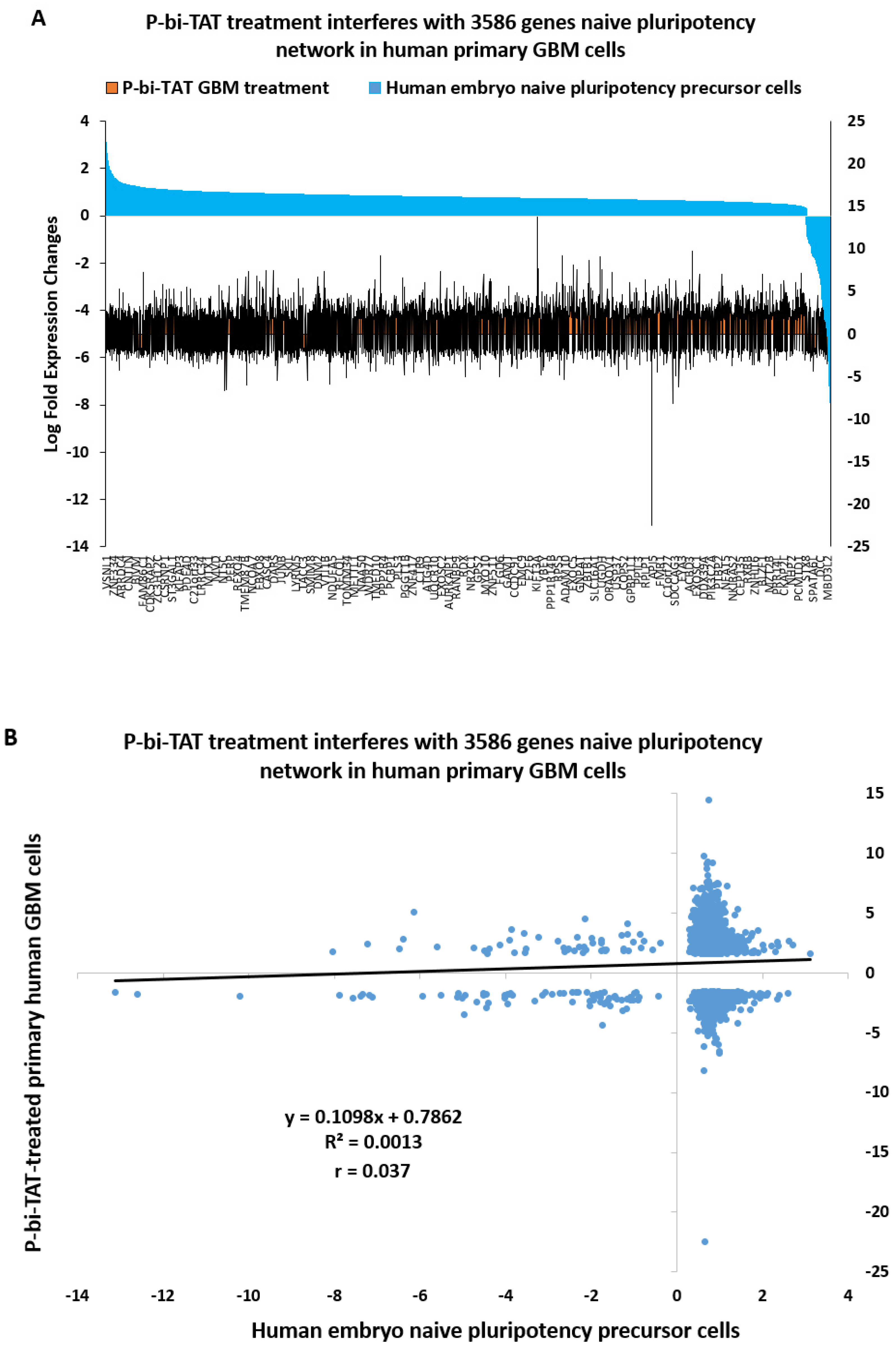
2.8. Naïve Pluripotency Network Genes of Human Preimplantation Embryo Comprise a Marked Majority of the P-bi-TAT Target Genes in Human GBM Cells
3. Discussion
4. Materials and Methods
4.1. P-bi-TAT
4.2. Cells and Cell Culture Conditions; Treatment with P-bi-TAT
4.3. Cell Proliferation Assay
4.4. Analysis of the P-bi-TAT Effects on Angiogenesis
4.5. Microarray
5. Conclusions
- Energy metabolism gene expression pathways represent an intrinsic component of stemness and cancer survival networks engaged in malignant cells;
- The association between cancer cells’ stemness state, survival networks and energy metabolism pathways revealed by thyrointegrin antagonist actions was observed in primary GBM cells and appears less evident in cancer cells adapted to in vitro cell culture conditions;
- This apparent dichotomy likely reflects different states of cancer cells’ adaptations to strikingly distinct in vivo and in vitro microenvironmental conditions favoring deployments of different energy metabolism mechanisms. Some important examples of such microenvironmental factors include availability of nutrients, degrees of oxygen accessibility and milieu acidification reaching extreme hypoxic and acidic conditions in vivo;
- In addition to fundamental and mechanistic considerations, documented effects of P-bi-TAT on gene expression of cancer stemness, survival and energy metabolism networks highlight a powerful therapeutic interference opportunity with growth and survival of malignant cells.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [Green Version]
- Davis, P.J.; Glinsky, G.V.; Lin, H.-Y.; Leith, J.T.; Hercbergs, A.; Tang, H.-Y.; Ashur-Fabian, O.; Incerpi, S.; Mousa, S.A. Cancer cell gene expression modulated from plasma membrane integrin αvβ3 by thyroid hormone and nanoparticulate tetrac. Front. Endocrinol. 2014, 5, 240. [Google Scholar]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef]
- Davis, P.J.; Mousa, S.A.; Lin, H.Y. Nongenomic actions of thyroid hormone: The integrin component. Physiol. Rev. 2021, 101, 319–352. [Google Scholar] [CrossRef]
- Davis, P.J.; Tang, H.Y.; Hercbergs, A.; Lin, H.Y.; Keating, K.A.; Mousa, S.A. Bioactivity of thyroid hormone analogs at cancer cells. Front. Endocrinol. 2018, 9, 739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousa, S.A.; Glinsky, G.V.; Lin, H.Y.; Ashur-Fabian, O.; Hercbergs, A.; Keating, K.A.; Davis, P.J. Contributions of thyroid hormone to cancer metastasis. Biomedicines 2018, 6, 89. [Google Scholar] [CrossRef] [Green Version]
- Rajabi, M.; Godugu, K.; Sudha, T.; Bharali, D.J.; Mousa, S.A. Triazole modified tetraiodothyroacetic acid conjugated to polyethylene glycol: High affinity thyrointegrin αvβ3 antagonist with potent anticancer activities in glioblastoma multiforme. Bioconjugate Chem. 2019, 30, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Glinsky, G.V.; Thangirala, S.; Lin, H.-Y.; Mousa, S.A. Abstract A48: Regulation of cancer cell metabolism via the thyroid hormone analogue receptor on integrin αvβ3: Actions of P-bi-TAT (tetrac-PEG) at the receptor. In Proceedings of the AACR Metabolism and Cancer Conference, New York, NY, USA, 28 September–1 October 2018. [Google Scholar]
- Shi, Y.; Kyun Lin, S.; Liang, Q.; Iyer, S.V.; Wang, H.-Y.; Wang, Z.; Xuanhua, X.; Sun, D.; Chen, Y.-J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylate inhibitor that targets glibolastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Yalcin, M.; Bharali, D.J.; Lansing, L.; Dyskin, E.; Mousa, S.S.; Hercbergs, A.; Davis, F.B.; Davis, P.J.; Mousa, S.A. Tetraidothyroacetic acid (tetrac) and tetrac nanoparticles inhibit growth of human renal cell carcinoma xenografts. Anticancer Res. 2009, 29, 3825–3831. [Google Scholar]
- Mousa, S.A.; Yalcin, M.; Bharali, D.J.; Meng, R.; Tang, H.Y.; Lin, H.Y.; Davis, F.B.; Davis, P.J. Tetraiodothyroacetic acid and its nanoformulation inhibit thyroid hormone stimulation of non-small cell lung cancer cells in vitro and its growth in xenografts. Lung Cancer 2012, 76, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Bharali, D.J.; Yalcin, M.; Davis, P.J.; Mousa, S.A. Tetraiodothyroacetic acid-conjugated PLGA nanoparticles: A nanomedicine approach to treat drug-resistant breast cancer. Nanomedicine 2013, 8, 1943–1954. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, M.; Lin, H.Y.; Sudha, T.; Bharali, D.J.; Meng, R.; Tang, H.Y.; Davis, F.B.; Stain, S.C.; Davis, P.J.; Mousa, S.A. Response of human pancreatic cancer cell xenografts to tetraiodothyroacetic acid nanoparticles. Horm. Cancer 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Sudha, T.; Bharali, D.J.; Sell, S.; Darwish, N.H.E.; Davis, P.J.; Mousa, S.A. Nanoparticulate tetrac inhibits growth and vascularity of glioblastoma xenografts. Horm. Cancer 2017, 8, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Mousa, S.A.; Sudha, T.; Godugu, K.; Rajabi, M.; Sell, S.; Davis, P.J. Abstract 1300: Novel thyrointegrin αvβ3 antagonist in the treatment of glioblastoma multiforme. Cancer Res. 2019, 79, 1300. [Google Scholar]
- Lo, H.W. EGFR-targeted therapy in malignant glioma: Novel aspects and mechanisms of drug resistance. Curr. Mol. Pharmacol. 2010, 3, 37–52. [Google Scholar] [CrossRef]
- Addeo, R.; Zappavigm, S.; Parlato, C.; Caraglia, M. Erlotinib: Eary clinical development in brain cancer. Expert Opin. Investig. Drugs 2014, 23, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.N. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Barash, R.; Asna, N.; Schaffer, P.; Francis, N.; Scaffner, M. Glioblastoma multiforme, diagnosis and treatment. Curr. Med. Chem. 2017, 24, 3002–3009. [Google Scholar]
- Lang, H.L.; Hu, G.W.; Zhang, B.; Kuang, W.; Chen, Y.; Wu, L.; Xu, G.H. Glioma cell enhance angiogenesis and inhibit endothelial cell apoptosis through the release of exosomes that contain long non-coding RNA CCAT2. Oncol. Rep. 2017, 38, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Zhang, Y.; Cheng, S.; Wu, Z.; Liu, F.; Zhang, J. CD133 positive U87 glioblastoma cells-derived exosomal microRNA in hyoxia-versus normoxia-microenvironment. J. Neuro-Oncol. 2017, 135, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xia, J.; Wang, T.; Li, W.; Song, Y.; Tan, G. Differentiation of human glioblastoma U87 cells into cholinergic neurons. Neurosci. Lett. 2019, 704, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Romanenko, M.V.; Dolgova, E.V.; Osipov, I.D.; Ritter, G.S.; Sizova, M.S.; Proskurina, A.S.; Efremov, Y.R.; Bayborodin, S.I.; Potter, E.A.; Taranov, O.S.; et al. Oncolytic effect of adenovirus serotypes and 6against U87 glioblastoma cancer stem cells. Anticancer Res. 2019, 39, 6073–6086. [Google Scholar] [CrossRef] [PubMed]
- Monnier, A.; Boniface, R.; Bouvet, R.; Etchverry, A.; Aubry, M.; Avril, T.; Quillien, V.; Chevet, E.; Mosser, J. The expression of EMX2 leads to cell cycle arrest in glioblastoma cell line. BMC Cancer 2018, 18, 1213. [Google Scholar] [CrossRef]
- He, T.; Chittur, S.V.; Musah, R.A. Impact on glioblastoma U87 cell gene expression of a carborane cluster-bearing amino acid: Implications for carborane toxicity in mammalian cells. Chem. Neurosci. 2019, 10, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Riabovol, O.O.; Tsymbal, D.O.; Minchenko, D.O.; Lebid-Biletska, K.M.; Sliusar, M.Y.; Rudnytsk, O.V.; Michenko, O.H. Effect of glucose deprivation on the expression of genes encoding glucocorticoid receptor and some related factors in ERN1-knockdown U87 glioma cells. Endocr. Regul. 2019, 53, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Debreli-Coskun, M.; Sudha, T.; Bharali, D.J.; Celikler, S.; Davis, P.J.; Mousa, S.A. αvβ3 Integrin antagonists emhance chemotherapy response in an orthotopic pancreatic cancer model. Front. Pharmacol. 2020, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Statis Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Glinsky, G.V.; Glinskii, A.B.; Stephenson, A.J.; Hoffman, R.N.; Gerald, W.L. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Investig. 2004, 113, 913–923. [Google Scholar] [CrossRef]
- Glinsky, G.V.; Higashiyama, T.; Glinskii, A.B. Classification of human breast cancer using gene expression profiling as a component of the survival predictor algorithm. Clin. Cancer Res. 2004, 10, 2272–2283. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef] [Green Version]
- Glinsky, G.V. Activation of endogenous human stem cell-associated retroviruses (SCARs) and therapy-resistant phenotypes of malignant tumors. Cancer Lett. 2016, 376, 347–359. [Google Scholar] [CrossRef]
- Glinsky, G.V. Single cell genomics reveals activation signatures of endogenous SCAR’s networks in aneuploid human embryos and clinically intractable malignant tumors. Cancer Lett. 2016, 381, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V. Genomics-Guided Drawing of Molecular and Pathophysiological Components of Malignant Regulatory Signatures Reveals a Pivotal Role in Human Diseases of Stem Cell-Associated Retroviral Sequences and Functionally-Active hESC Enhancers. Front. Oncol. 2021, 11, 974. [Google Scholar] [CrossRef]
- Kieseling, M.K.; Curioni-Fontecedro, A.; Samaras, P.; Atrott, K.; Cosin-Roger, J.; Lang, S.; Scharl, M.; Rogler, G. Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget 2015, 6, 42183–42196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Cha, H.; Kim, H.; Lee, J.H.; Park, J.W. Amelioration of late-onset hepatic steatosis in IDH2-deficient mice. Free Radic. Res. 2017, 51, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Tischler, J.; Gruhn, W.H.; Reid, J.; Allgeyer, E.; Buettner, F.; Marr, C.; Theis, F.; Simons, B.D.; Wernisch, L.; Surani, M.A. Metabolic regulation of pluripotency and germ cell fate throukgh alpha-ketoglutarate. EMBO J. 2018, 38, e99518. [Google Scholar]
- Magnol, L.; Chevallier, M.C.; Nalesso, V.; Retif, S.; Fuchs, H.; Klempt, M.; Pereira, P.; Riottot, M.; Andrzejewski, S.; Doan, B.T.; et al. KIT is required for hepatic function during mouse post-natal development. BMC Dev. Biol. 2007, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Jin, C.; Qin, L.; Shi, Y.; Candas, D.; Fan, M.; Lu, C.L.; Vaughan, A.T.; Shen, R.; Wu, L.S.; Liu, R.; et al. CDK4-mediated MnSOD activation and mitochondrial homeostasis in radioadaptive protection. Free Radic. Biol. Med. 2015, 81, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Smeitk, J.A.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architeccture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Glinskii, A.B.; Glinsky, G.V.; Lin, H.Y.; Tang, H.Y.; Sun, M.; Davis, F.B.; Luidens, M.K.; Mousa, S.A.; Hercbergs, A.H.; Davis, P.J. Modification of survival pathway gene expression in human breast cancer cells by tetraiodothyroacetic acid (tetrac). Cell Cycle 2009, 8, 3562–3570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalcin, M.; Dyskin, E.; Lansing, L.; Bharali, D.J.; Mousa, S.S.; Bridoux, A.; Hercbergs, A.H.; Lin, H.Y.; Davis, F.B.; Glinsky, G.V.; et al. Tetraiodothyroacetic acid (tetrac) and nanopartiulate tetrac arrest growth of medullary carcinoma of the thyroid. J. Clinl. Endocrinol. Metab. 2010, 95, 1972–1980. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chin, Y.T.; Nana, A.W.; Shih, Y.J.; Lai, H.U.; Tang, H.Y.; Leinung, M.; Mousa, S.A.; Davis, P.J. Actions of l-thyroxine and Nano-diamino-tetrac (Nanotetrac) on PD-L1 in cancer cells. Steroids 2016, 114, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, G.V. Human-specific genomic features of pluripotency regulatory networks link NANOG with fetal and adult brain development. bioRxiv 2017. bioRxiv:022913. [Google Scholar]
- Glinsky, G.; Durruthy-Durruthy, J.; Wossidlo, M.; Grow, E.J.; Weirather, J.L.; Au, K.F.; Wysocka, J.; Sebastiano, V. Single cell expression analysis of primate-specific retroviruses-derived HPAT lincRNAs in viable human blastocysts identifies embryonic cells co-expressing genetic markers of multiple lineages. Heliyon 2018, 4, e00667. [Google Scholar] [CrossRef]
- Glinsky, G.V. Contribution of transposable elements and distal enhancers to evolution of human-specific features of interphase chromatin architecture in embryonic stem cells. Chromosome Res. 2018, 26, 61–84. [Google Scholar] [CrossRef]
- Glinsky, G.; Barakat, T.S. The evolution of Great Apes has shaped the functional enhancers’ landscape in human embryonic stem cells. Stem Cell Res. 2019, 37, 101456. [Google Scholar] [CrossRef]
- Ahluwlia, M.S.; Chang, S.M. Medical therapy of, gliomas. J. Neuro-Oncol. 2014, 119, 503–512. [Google Scholar] [CrossRef]
- Glaser, T.; Han, I.; Wu, L.; Zeng, X. Targeted nanotechnology in glioblastoma multiforme. Front. Pharmacol. 2019, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Munivandi, P.; Maekawa, T.; Kumar, D.S. Vesicular systems employing natural substances as proising drug candidates for MMP inhibition in glioblastoma: Nanotechnological approach. Int. J. Pharm. 2018, 551, 339–361. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.T.; He, Z.R.; Chen, C.I.; Chu, H.C.; Ho, Y.; Su, P.Y.; Yang, Y.S.H.; Wang, K.; Shih, Y.J.; Chen, Y.R.; et al. Tetrac and NDAT induce anti-proliferation via integrin αvβ3 in colorectal cancers with different K-RAS status. Front. Endocrinol. 2019, 10, 130. [Google Scholar] [CrossRef] [Green Version]
- Shinderman-Maman, E.; Cohen, K.; Weingarten, C.; Nabriski, D.; Twito, O.; Baraf, L.; Herbergs, A.; Davis, P.J.; Werner, H.; Ellis, M. The thyroid hormone-αvβ3 integrin axis in ovarian cancer: Regulation of gene transcription and MAPK-dependent proliferation. Oncogene 2016, 35, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chin, Y.T.; Ho, Y.; Chou, S.Y.; Sh Yang, Y.C.; Nana, A.W.; Su, K.W.; Lim, Y.T.; Wang, K.; Lee, S.Y.; et al. Nano-diamino-tetrac (NDAT) inhibits PD-L1 expression which is essential for proliferation in oral cancer cells. Food Chem. Toxicol. 2018, 120, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Landersdorfer, C.B.; London, D.; Meng, R.; Lim, C.U.; Lin, C.; Lin, S.; Tang, H.Y.; Brown, D.; Van Scoy, B.; et al. Pharmacodynamic modeling of anti-cancer activity of tetraiodothyroacetic acid in a perfused cell culture system. PLoS Comput. Biol. 2011, 7, e1001073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, P.J.; Incerpi, S.; Lin, H.Y.; Sudha, T.; Mousa, S.A. Thyroid hormone and p-glycoprotein in cancer cells. Biomed. Res. Int. 2015, 2015, 168427. [Google Scholar] [CrossRef]
- Eguchi, Y.; Shimizu, S.; Tsujimpto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1831–1840. [Google Scholar]
- Singh, S.; Khar, A. Differential gene expression during apoptosis induced by a serum factor: Role of mitochondrial F0-F1 ATP synthase complex. Apoptosis 2005, 10, 1469–1482. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V. Transposable Elements and DNA Methylation Create in Embryonic Stem Cells Human-Specific Regulatory Sequences Associated with Distal Enhancers and Noncoding RNAs. Genome Biol. Evol. 2015, 7, 1432–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, G.V. Mechanistically Distinct Pathways of Divergent Regulatory DNA Creation Contribute to Evolution of Human-Specific Genomic Regulatory Networks Driving Phenotypic Divergence of Homo sapiens. Genome Biol. Evol. 2016, 8, 2774–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, G.V. A Catalogue of 59,732 Human-Specific Regulatory Sequences Reveals Unique-to-Human Regulatory Patterns Associated with Virus-Interacting Proteins, Pluripotency, and Brain Development. DNA Cell Biol. 2020, 39, 126–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, G.V. Impacts of genomic networks governed by human-specific regulatory sequences and genetic loci harboring fixed human-specific neuro-regulatory single nucleotide mutations on phenotypic traits of modern humans. Chromosome Res. 2020, 28, 331–353. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification Category | Number of Differentially Regulated Genes | Number of Cancer Survival Genes | Percent of Cancer Survival Genes | p Value |
|---|---|---|---|---|
| Pancreatic cancer (SUIT2) | 1293 | 860 | 67 | 1.47 × 10−19 |
| Top upregulated (p = 0.001) | 50 | 40 | 80 | 0.000124 |
| Top downregulated (p = 0.001) | 15 | 12 | 80 | 0.030497 |
| Top differentially regulated (p = 0.001) | 65 | 52 | 80 | 1.32 × 10−5 |
| Glioblastoma multiforme (GBM) | 5362 | 3403 | 63 | 1.91 × 10−52 |
| Top upregulated (6-fold) | 66 | 47 | 71 | 0.002467 |
| Top downregulated (2.5-fold) | 106 | 64 | 60 | 0.039735 |
| Top differentially regulated | 172 | 111 | 65 | 0.002074 |
| Consensus (SUIT2 and GBM) | 737 | 501 | 68 | 2.5 × 10−14 |
| Top upregulated (4.9-fold) | 68 | 57 | 84 | 2.91 × 10−7 |
| Top downregulated (2.5-fold) | 61 | 43 | 70 | 0.004617 |
| Top differentially regulated | 129 | 100 | 78 | 3.88 × 10−8 |
| Classification Category | Downregulated Genes |
|---|---|
| Electron transport chain | ATP5A1, ATP5I, COX6B1, ATP5G2, NDUFA8, NDUFA3, NDUFV2, NDUFA6, NDUFA2, COX5A, NDUFS7, COX6A1, COX4I1, SLC25A6, NDUFB3, ATP5G1, COX7A2, ND6, NDUFAB1, COX7B, NDUFB7, UQCRC1, COX5B, COX8A, NDUFV1, ATP5G3, SURF1, NDUFB2, NDUFS2, ATP5D, NDUFV3, NDUFA10, UCP2, NDUFS8, NDUFB8 |
| Cytoplasmic ribosomal proteins | RPL10A, RPL8, RPL9, RPLP2, RPLP1, RPL35, RPL7A, RPL13, RPL14, RPL18A, RPL18, RPL19, RPL21, RPL27, RPL28, RPL29, RPL32, RPL39, UBA52, RPL41, RPL36A, RPS3, RPS9, RPS5, RPS15A, RPS16, RPS20, RPS14, RPS29, RPS11, RPS15, RPS7, RPS8, RPS10, RPS19, RPS26, RPS27, RPS27A, RPS28, FAU, RPLP0, RPS6KA1, RPL11, RPL10, RPL30, RPS2, RPS6KB2 |
| Oxidative phosphorylation | ATP5A1, ATP5D, ATP5G2, ATP5G1, ATP5G3, ATP5I, NDUFA11, NDUFS7, NDUFA2, ND6, NDUFA8, NDUFS2, NDUFS8, NDUFB2, NDUFV2, NDUFV3 |
| Metabolism of carbohydrates | SLC25A1, PCK1, SLC25A10, GALK1, GALT, PGLS, SLC37A4, AKR1B1, AKR1A1 |
| Glucose metabolism | SLC25A1, PCK1, SLC25A10, SLC37A4 |
| Fatty acyl-CoA and cholesterol biosynthesis | SLC25A1, PPT2, SLC27A3, FDPS, MVD, DHCR7, PMVK, FDFT1, MVK |
| Globo sphingolipid metabolism | ST3GAL1, ST6GALNAC4, ST6GALNAC6, ST6GAL1 |
| Biogenic amine synthesis | DDC, ACHE, COMT |
| Pathway | Cancer Model | Number of Genes | p-Value | Cancer Model | Number of Genes | p-Value |
|---|---|---|---|---|---|---|
| VEGFA-VEGFR2 signaling pathway | GBM | 85 | 0.011056 | SUIT2 | 29 | 0.00245 |
| Androgen receptor signaling pathway | GBM | 43 | 0.000046 | SUIT2 | 13 | 0.00843 |
| Brain-derived neurotrophic factor (BDNF) signaling pathway | GBM | 54 | 0.020347 | SUIT2 | 19 | 0.00678 |
| Deubiquitination | GBM | 13 | 0 | SUIT2 | 4 | 0 |
| Endoderm differentiation | GBM | 53 | 0.025638 | SUIT2 | 17 | 0.02808 |
| Focal adhesion | GBM | 69 | 0.011855 | SUIT2 | 24 | 0.00308 |
| Gastric cancer network 2 | GBM | 15 | 0.027934 | SUIT2 | 7 | 0.00467 |
| Human thyroid-stimulating hormone (TSH) signaling pathway | GBM | 30 | 0.002383 | SUIT2 | 10 | 0.01235 |
| IL-6 signaling pathway | GBM | 22 | 0.001737 | SUIT2 | 9 | 0.00193 |
| Integrin-mediated cell adhesion | GBM | 40 | 0.013987 | SUIT2 | 14 | 0.0085 |
| Interleukin-11 signaling pathway | GBM | 27 | 0.000004 | SUIT2 | 8 | 0.00831 |
| MAPK signaling pathway | GBM | 16 | 0.002772 | SUIT2 | 21 | 0.00768 |
| Olfactory receptor activity | GBM | 47 | 0 | SUIT2 | 6 | 4 × 10−6 |
| Signaling of hepatocyte growth factor (HGF) receptor | GBM | 16 | 0.020177 | SUIT2 | 6 | 0.02432 |
| TCF-dependent signaling in response to WNT | GBM | 14 | 0 | SUIT2 | 9 | 0.00539 |
| TGF-beta signaling pathway | GBM | 61 | 0.000019 | SUIT2 | 17 | 0.01388 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glinsky, G.V.; Godugu, K.; Sudha, T.; Rajabi, M.; Chittur, S.V.; Hercbergs, A.A.; Mousa, S.A.; Davis, P.J. Effects of Anticancer Agent P-bi-TAT on Gene Expression Link the Integrin Thyroid Hormone Receptor to Expression of Stemness and Energy Metabolism Genes in Cancer Cells. Metabolites 2022, 12, 325. https://doi.org/10.3390/metabo12040325
Glinsky GV, Godugu K, Sudha T, Rajabi M, Chittur SV, Hercbergs AA, Mousa SA, Davis PJ. Effects of Anticancer Agent P-bi-TAT on Gene Expression Link the Integrin Thyroid Hormone Receptor to Expression of Stemness and Energy Metabolism Genes in Cancer Cells. Metabolites. 2022; 12(4):325. https://doi.org/10.3390/metabo12040325
Chicago/Turabian StyleGlinsky, Gennadi V., Kavitha Godugu, Thangirala Sudha, Mehdi Rajabi, Sridar V. Chittur, Aleck A. Hercbergs, Shaker A. Mousa, and Paul J. Davis. 2022. "Effects of Anticancer Agent P-bi-TAT on Gene Expression Link the Integrin Thyroid Hormone Receptor to Expression of Stemness and Energy Metabolism Genes in Cancer Cells" Metabolites 12, no. 4: 325. https://doi.org/10.3390/metabo12040325