
Advances in Metabolomics-Driven Diagnostic Breeding and Crop Improvement

 ,
,  ,
, 
Abstract
:1. Introduction
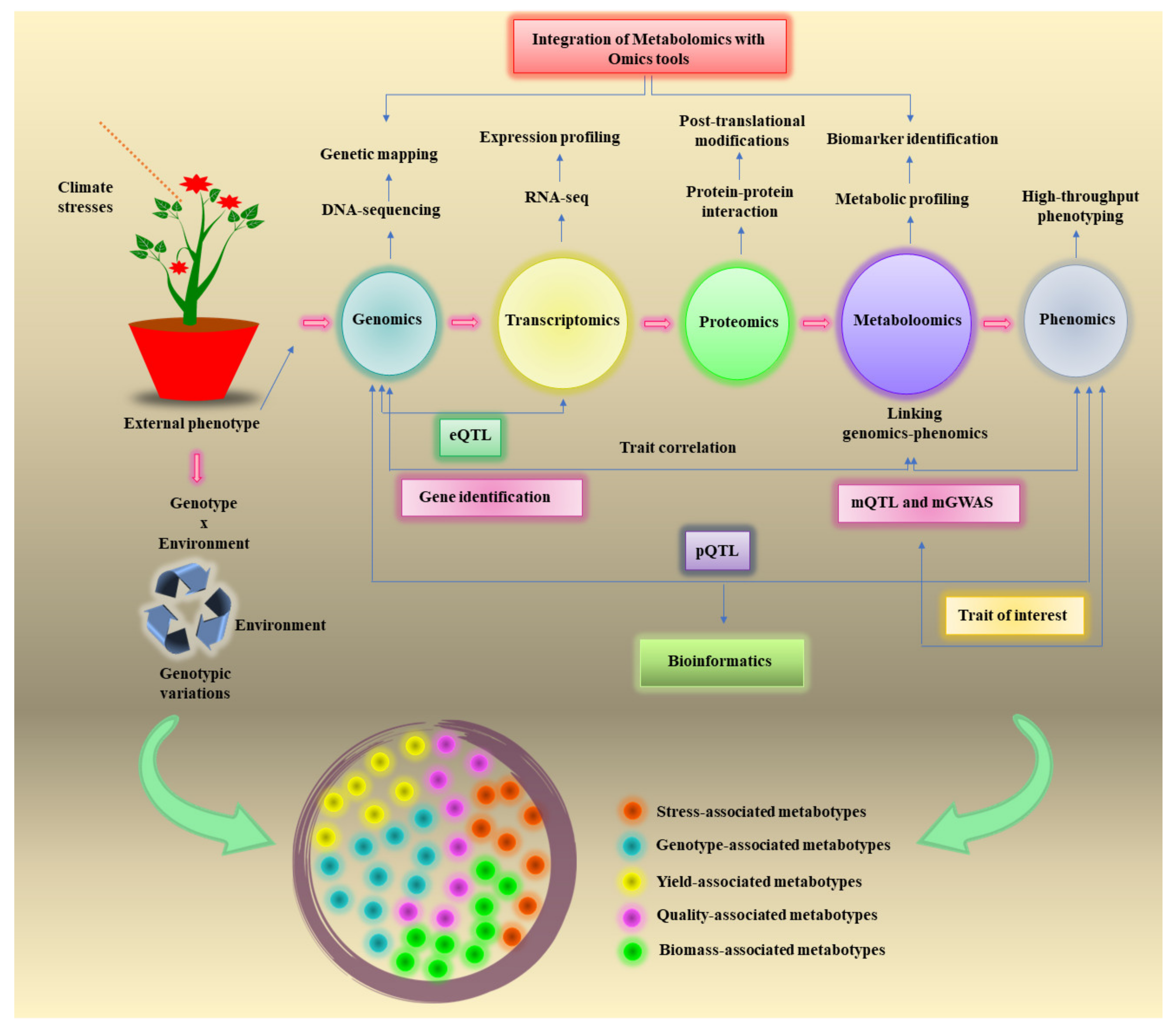
2. Linking Genomics to Phenomics through Metabolomics-Assisted Breeding
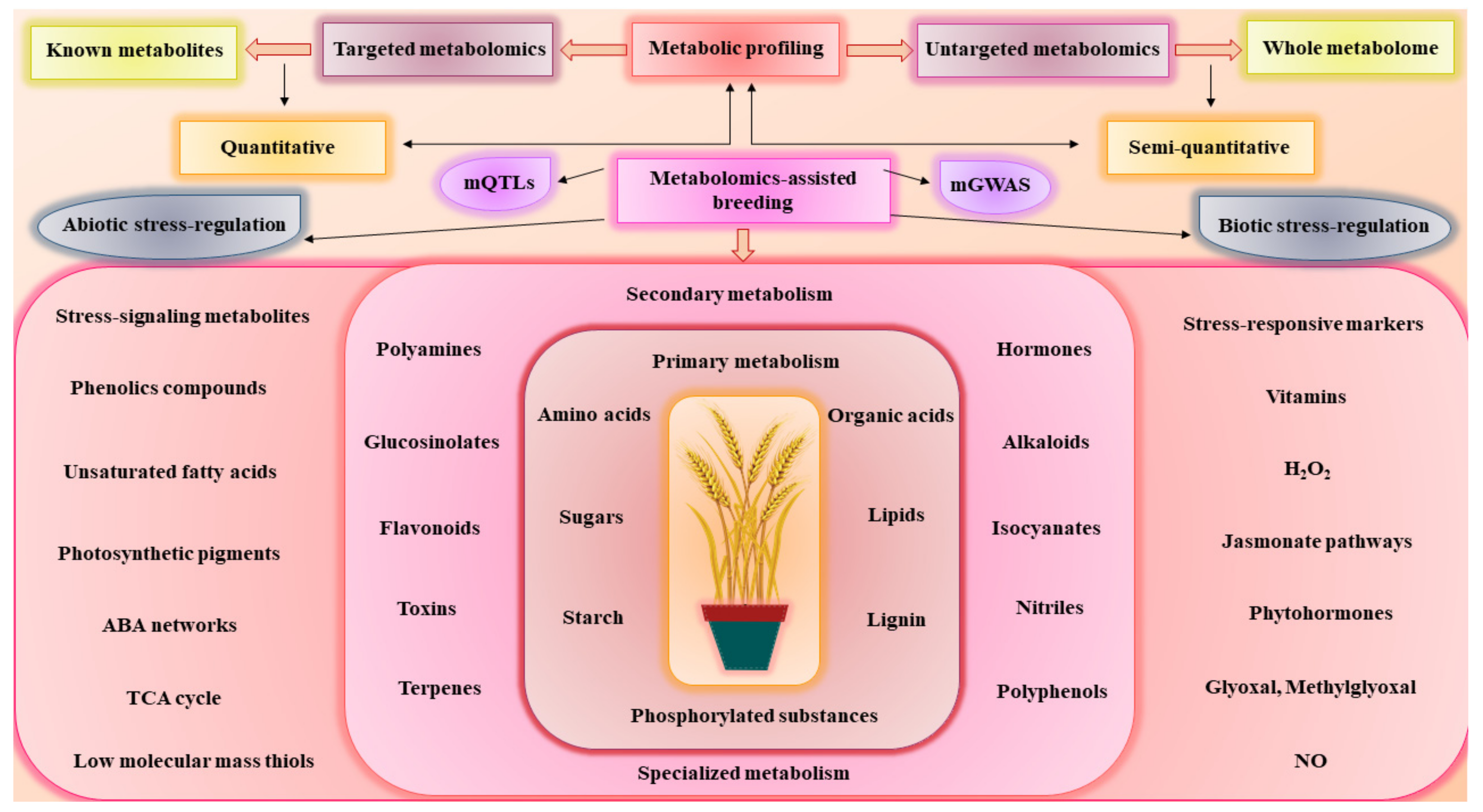
3. Metabolomics-Assisted Breeding for Agronomics Traits
4. Integration of Metabolomics with OMICS Tools for Climate Resilience
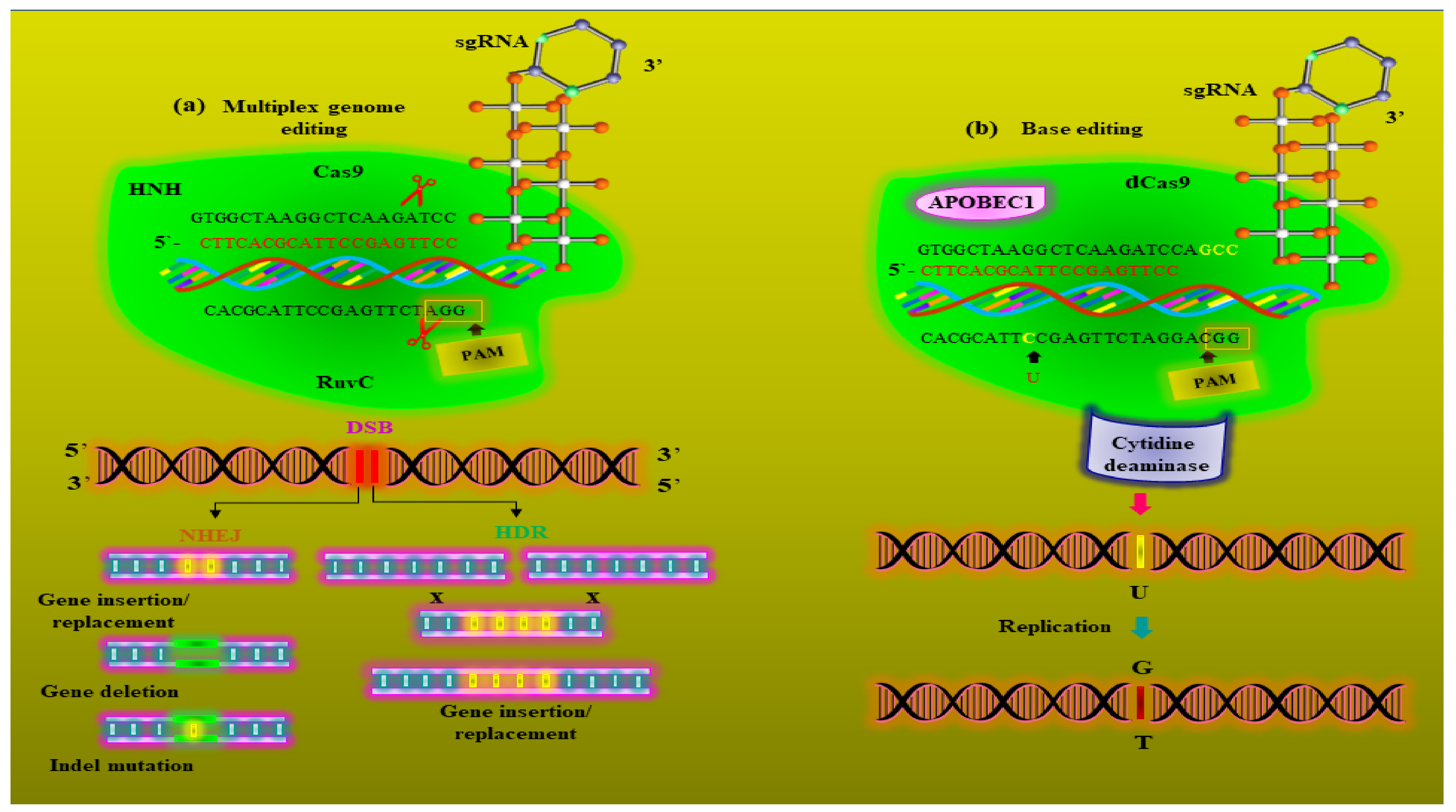
5. Metabolic Engineering and Metabolic Editing
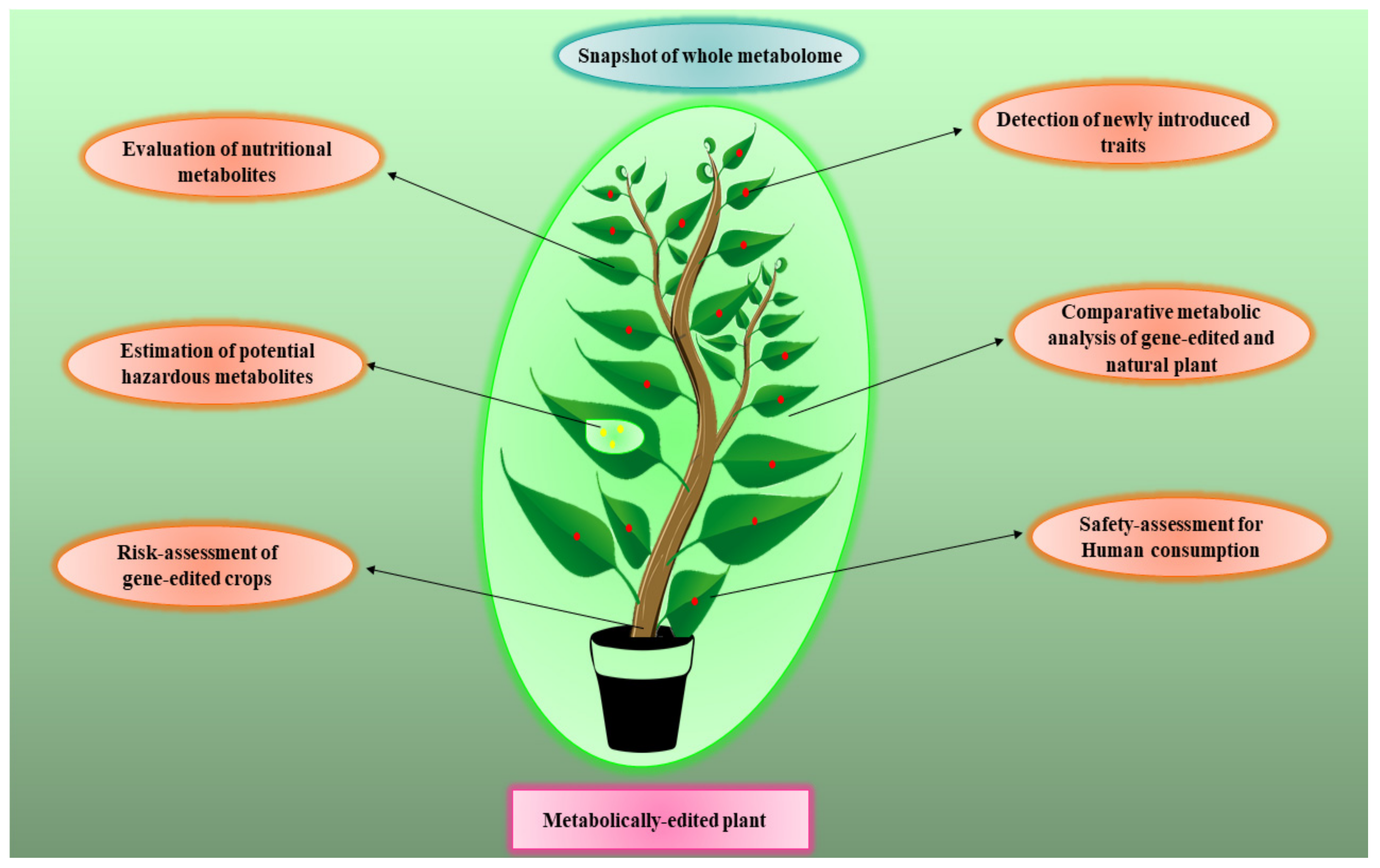
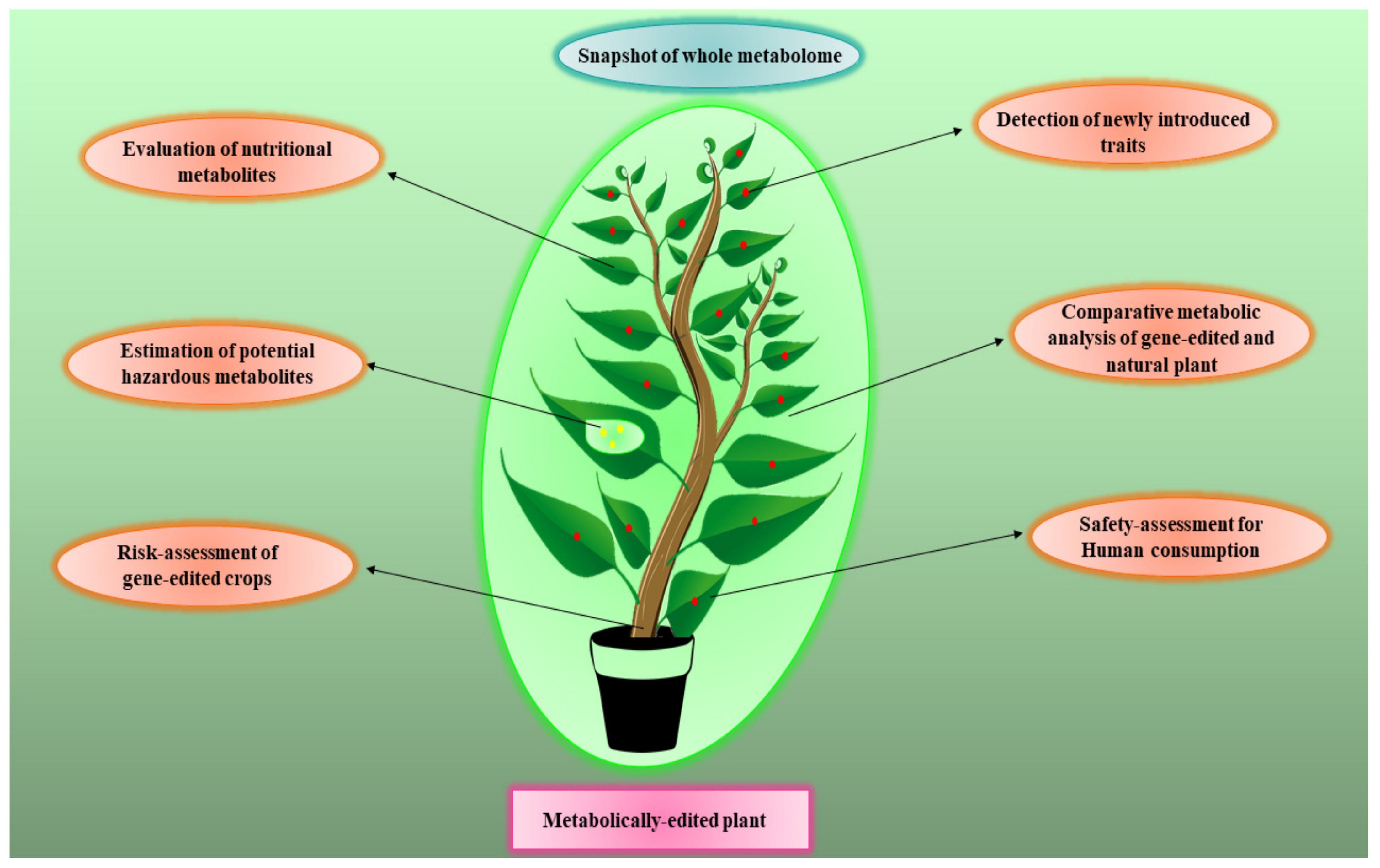
6. Metabolomics for Risk Assessment of Gene-Edited Crops
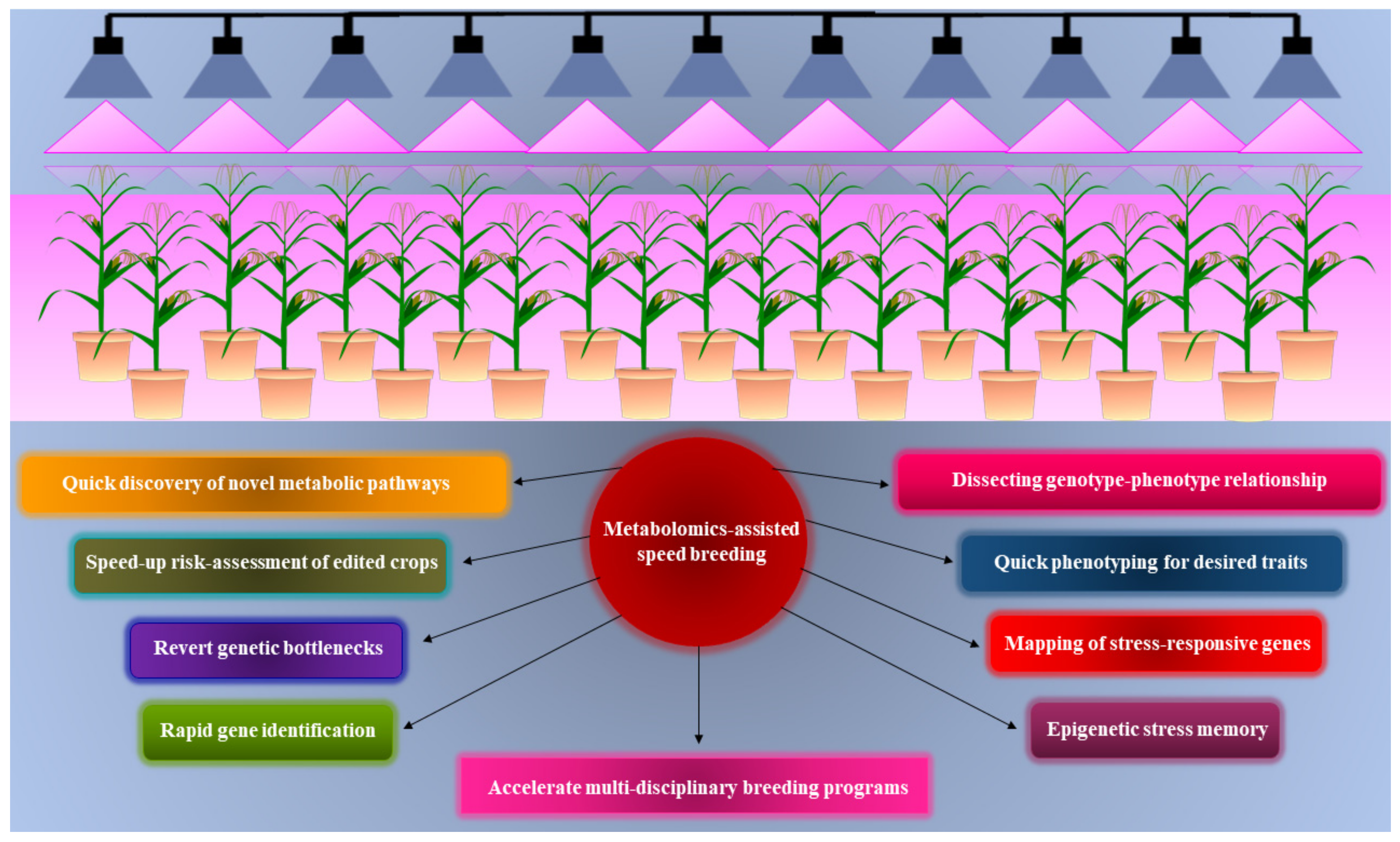
7. Metabolomics-Assisted Speed Breeding
8. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- FAO. State of Food Security and Nutrition in the World. Available online: https://www.fao.org/publications/sofi/2021/en/ (accessed on 15 January 2022).
- Raza, A.; Razzaq, A.; Mehmood, S.S.; Zou, X.; Zhang, X.; Lv, Y.; Xu, J. Impact of climate change on crops adaptation and strategies to tackle its outcome: A review. Plants 2019, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzaq, A.; Kaur, P.; Akhter, N.; Wani, S.H.; Saleem, F. Next-generation breeding strategies for climate-ready crops. Front. Plant Sci. 2021, 12, 620420. [Google Scholar] [CrossRef]
- Aggarwal, P.; Vyas, S.; Thornton, P.; Campbell, B.M.; Kropff, M. Importance of considering technology growth in impact assessments of climate change on agriculture. Glob. Food Secur. 2019, 23, 41–48. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Foito, A.; Stewart, D. Metabolomics: A high-throughput screen for biochemical and bioactivity diversity in plants and crops. Curr. Pharm. Des. 2018, 24, 2043–2054. [Google Scholar] [CrossRef]
- Piasecka, A.; Kachlicki, P.; Stobiecki, M. Analytical methods for detection of plant metabolomes changes in response to biotic and abiotic stresses. Int. J. Mol. Sci. 2019, 20, 379. [Google Scholar] [CrossRef] [Green Version]
- Razzaq, A.; Sadia, B.; Raza, A.; Khalid Hameed, M.; Saleem, F. Metabolomics: A way forward for crop improvement. Metabolites 2019, 9, 303. [Google Scholar] [CrossRef] [Green Version]
- Sulpice, R. Closing the yield gap: Can metabolomics be of help? J. Exp. Bot. 2020, 71, 461–464. [Google Scholar] [CrossRef]
- Wani, S.H. Recent Approaches in Omics for Plant Resilience to Climate Change; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Fernie, A.R.; Stitt, M. On the discordance of metabolomics with proteomics and transcriptomics: Coping with increasing complexity in logic, chemistry, and network interactions scientific correspondence. Plant Physiol. 2012, 158, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Scossa, F.; Alseekh, S.; Fernie, A.R. Integrating multi-omics data for crop improvement. J. Plant Physiol. 2021, 257, 153352. [Google Scholar] [CrossRef]
- Moreno-Risueno, M.A.; Busch, W.; Benfey, P.N. Omics meet networks—using systems approaches to infer regulatory networks in plants. Curr. Opin. Plant Biol. 2010, 13, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.; Robards, K. Metabolomics: The greatest omics of them all? Anal. Chem. 2006, 78, 7954–7958. [Google Scholar] [CrossRef] [PubMed]
- Villate, A.; San Nicolas, M.; Gallastegi, M.; Aulas, P.-A.; Olivares, M.; Usobiaga, A.; Etxebarria, N.; Aizpurua-Olaizola, O. Metabolomics as a prediction tool for plants performance under environmental stress. Plant Sci. 2021, 303, 110789. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Schauer, N. Metabolomics-assisted breeding: A viable option for crop improvement? Trends Genet. 2009, 25, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Hameed, M.K.; Umar, W.; Razzaq, A.; Aziz, T.; Maqsood, M.A.; Wei, S.; Niu, Q.; Huang, D.; Chang, L. Differential Metabolic Responses of Lettuce Grown in Soil, Substrate and Hydroponic Cultivation Systems under NH4+/NO3− Application. Metabolites 2022, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Fernie, A.R. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.; Babar, M.A.; Khan, N.; Guo, J.; Khan, J.; Islam, S.; Shrestha, S.; Shahi, D. Comparative metabolomic profiling in the roots and leaves in contrasting genotypes reveals complex mechanisms involved in post-anthesis drought tolerance in wheat. PLoS ONE 2019, 14, e0213502. [Google Scholar] [CrossRef]
- Rosato, A.; Tenori, L.; Cascante, M.; Carulla, P.R.D.A.; Dos Santos, V.A.M.; Saccenti, E. From correlation to causation: Analysis of metabolomics data using systems biology approaches. Metabolomics 2018, 14, 37. [Google Scholar] [CrossRef] [Green Version]
- Weckwerth, W. Metabolomics: An integral technique in systems biology. Bioanalysis 2010, 2, 829–836. [Google Scholar] [CrossRef]
- Weckwerth, W. Toward a unification of system-theoretical principles in biology and ecology—the stochastic lyapunov matrix equation and its inverse application. Front. Appl. Math. Stat. 2019, 5, 29. [Google Scholar] [CrossRef]
- Feng, L.; Wang, C.; Yang, X.; Jiao, Q.; Yin, Y. Transcriptomics and metabolomics analyses identified key genes associated with sugar and acid metabolism in sweet and sour pomegranate cultivars during the developmental period. Plant Physiol. Biochem. 2022, 181, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Luo, J. Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J. 2019, 97, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Yadav, C.B.; Srivastava, R.K.; Gangashetty, P.I.; Yadav, R.; Mur, L.A.; Yadav, R.S. Metabolite Diversity and Metabolic Genome-Wide Marker Association Studies (mGWAS) for Health Benefiting Nutritional Traits in Pearl Millet Grains. Cells 2021, 10, 3076. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, W.; Peng, M.; Gong, L.; Gao, Y.; Wan, J.; Wang, S.; Shi, L.; Zhou, B.; Li, Z. Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat. Commun. 2016, 7, 12767. [Google Scholar] [CrossRef] [PubMed]
- Matros, A.; Liu, G.; Hartmann, A.; Jiang, Y.; Zhao, Y.; Wang, H.; Ebmeyer, E.; Korzun, V.; Schachschneider, R.; Kazman, E. Genome–metabolite associations revealed low heritability, high genetic complexity, and causal relations for leaf metabolites in winter wheat (Triticum aestivum). J. Exp. Bot. 2017, 68, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Kremling, K.A.; Bandillo, N.; Richter, A.; Zhang, Y.K.; Ahern, K.R.; Artyukhin, A.B.; Hui, J.X.; Younkin, G.C.; Schroeder, F.C. Metabolome-scale genome-wide association studies reveal chemical diversity and genetic control of maize specialized metabolites. Plant Cell 2019, 31, 937–955. [Google Scholar] [CrossRef]
- Liang, X.; Liu, S.; Wang, T.; Li, F.; Cheng, J.; Lai, J.; Qin, F.; Li, Z.; Wang, X.; Jiang, C. Metabolomics-driven gene mining and genetic improvement of tolerance to salt-induced osmotic stress in maize. New Phytol. 2021, 230, 2355–2370. [Google Scholar] [CrossRef]
- Liu, J.Y.; Li, P.; Zhang, Y.W.; Zuo, J.F.; Li, G.; Han, X.; Dunwell, J.M.; Zhang, Y.M. Three-dimensional genetic networks among seed oil-related traits, metabolites and genes reveal the genetic foundations of oil synthesis in soybean. Plant J. 2020, 103, 1103–1124. [Google Scholar] [CrossRef]
- Wei, W.; Li, S.; Wang, Y.; Wang, B.; Fan, G.; Zeng, Q.; Zhao, F.; Xu, C.; Zhang, X.; Tang, T. Metabolome-Based Genome-Wide Association Study Provides Genetic Insights Into the Natural Variation of Foxtail Millet. Front. Plant Sci. 2021, 12, 665530. [Google Scholar] [CrossRef]
- Chen, J.; Hu, X.; Shi, T.; Yin, H.; Sun, D.; Hao, Y.; Xia, X.; Luo, J.; Fernie, A.R.; He, Z. Metabolite-based genome-wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnol. J. 2020, 18, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261.e12. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Zhu, A.; Jia, J.; Hu, X.; Chen, J.; Liu, W.; Ren, X.; Sun, D.; Fernie, A.R.; Cui, F. Metabolomics analysis and metabolite-agronomic trait associations using kernels of wheat (Triticum aestivum) recombinant inbred lines. Plant J. 2020, 103, 279–292. [Google Scholar] [CrossRef] [Green Version]
- Piasecka, A.; Sawikowska, A.; Kuczyńska, A.; Ogrodowicz, P.; Mikołajczak, K.; Krystkowiak, K.; Gudyś, K.; Guzy-Wróbelska, J.; Krajewski, P.; Kachlicki, P. Drought-related secondary metabolites of barley (Hordeum vulgare L.) leaves and their metabolomic quantitative trait loci. Plant J. 2017, 89, 898–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Templer, S.E.; Ammon, A.; Pscheidt, D.; Ciobotea, O.; Schuy, C.; McCollum, C.; Sonnewald, U.; Hanemann, A.; Förster, J.; Ordon, F. Metabolite profiling of barley flag leaves under drought and combined heat and drought stress reveals metabolic QTLs for metabolites associated with antioxidant defense. J. Exp. Bot. 2017, 68, 1697–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alseekh, S.; Tong, H.; Scossa, F.; Brotman, Y.; Vigroux, F.; Tohge, T.; Ofner, I.; Zamir, D.; Nikoloski, Z.; Fernie, A.R. Canalization of tomato fruit metabolism. Plant Cell 2017, 29, 2753–2765. [Google Scholar] [CrossRef]
- Alseekh, S.; Tohge, T.; Wendenberg, R.; Scossa, F.; Omranian, N.; Li, J.; Kleessen, S.; Giavalisco, P.; Pleban, T.; Mueller-Roeber, B. Identification and mode of inheritance of quantitative trait loci for secondary metabolite abundance in tomato. Plant Cell 2015, 27, 485–512. [Google Scholar] [CrossRef] [Green Version]
- Labadie, M.; Vallin, G.; Petit, A.; Ring, L.; Hoffmann, T.; Gaston, A.; Potier, A.; Schwab, W.; Rothan, C.; Denoyes, B. Metabolite quantitative trait loci for flavonoids provide new insights into the genetic architecture of strawberry (Fragaria× ananassa) fruit quality. J. Agric. Food Chem. 2020, 68, 6927–6939. [Google Scholar] [CrossRef]
- Li, K.; Wang, D.; Gong, L.; Lyu, Y.; Guo, H.; Chen, W.; Jin, C.; Liu, X.; Fang, C.; Luo, J. Comparative analysis of metabolome of rice seeds at three developmental stages using a recombinant inbred line population. Plant J. 2019, 100, 908–922. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, O.; Urrutia, M.; Bernillon, S.; Giauffret, C.; Tardieu, F.; Le Gouis, J.; Langlade, N.; Charcosset, A.; Moing, A.; Gibon, Y. Fortune telling: Metabolic markers of plant performance. Metabolomics 2016, 12, 158. [Google Scholar] [CrossRef] [Green Version]
- Suharti, W.S.; Nose, A.; Zheng, S.-H. Metabolomic study of two rice lines infected by Rhizoctonia solani in negative ion mode by CE/TOF-MS. J. Plant Physiol. 2016, 206, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Cuperlovic-Culf, M.; Wang, L.; Forseille, L.; Boyle, K.; Merkley, N.; Burton, I.; Fobert, P.R. Metabolic biomarker panels of response to fusarium head blight infection in different wheat varieties. PLoS ONE 2016, 11, e0153642. [Google Scholar] [CrossRef]
- Dhokane, D.; Karre, S.; Kushalappa, A.C.; McCartney, C. Integrated metabolo-transcriptomics reveals Fusarium head blight candidate resistance genes in wheat QTL-Fhb2. PLoS ONE 2016, 11, e0155851. [Google Scholar] [CrossRef] [Green Version]
- Shelp, B.J.; Bozzo, G.G.; Trobacher, C.P.; Zarei, A.; Deyman, K.L.; Brikis, C.J. Hypothesis/review: Contribution of putrescine to 4-aminobutyrate (GABA) production in response to abiotic stress. Plant Sci. 2012, 193, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Zhao, L.; Li, W.; Zhao, J.; Yan, J.; Ma, X.; Li, A.; Wang, H.; Kong, L. Integrated metabolo-transcriptomics and functional characterization reveals that the wheat auxin receptor TIR1 negatively regulates defense against Fusarium graminearum. J. Integr. Plant Biol. 2020, 63, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Van den Broeck, L.; Karre, S.; Choi, H.; Christensen, S.A.; Wang, G.F.; Jo, Y.; Cho, W.K.; Balint-Kurti, P. Analysis of the transcriptomic, metabolomic, and gene regulatory responses to Puccinia sorghi in maize. Mol. Plant Pathol. 2021, 22, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Karre, S.; Kumar, A.; Dhokane, D.; Kushalappa, A.C. Metabolo-transcriptome profiling of barley reveals induction of chitin elicitor receptor kinase gene (HvCERK1) conferring resistance against Fusarium graminearum. Plant Mol. Biol. 2017, 93, 247–267. [Google Scholar] [CrossRef]
- Pandey, V.; Singh, M.; Pandey, D.; Kumar, A. Integrated proteomics, genomics, metabolomics approaches reveal oxalic acid as pathogenicity factor in Tilletia indica inciting Karnal bunt disease of wheat. Sci. Rep. 2018, 8, 7826. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Q.; He, W.; He, T.; Wu, Q.; Miao, Y. Combined de novo transcriptome and metabolome analysis of common bean response to Fusarium oxysporum f. sp. phaseoli infection. Int. J. Mol. Sci. 2019, 20, 6278. [Google Scholar] [CrossRef] [Green Version]
- Yogendra, K.N.; Kushalappa, A.C. Integrated transcriptomics and metabolomics reveal induction of hierarchies of resistance genes in potato against late blight. Funct. Plant Biol. 2016, 43, 766–782. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Shi, L.; Jiao, Y.; Li, M.; Zhong, X.; Gu, F.; Liu, Q.; Xia, X.; Li, H. Metabolic responses to drought stress in the tissues of drought-tolerant and drought-sensitive wheat genotype seedlings. AoB Plants 2018, 10, ply016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzaq, A.; Guul, W.; Khan, M.S.; Saleem, F. Metabolomics: A powerful tool to study the complexity of wheat metabolome. Protein Pept. Lett. 2021, 28, 878–895. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xin, Z.; Yang, T.; Ma, X.; Zhang, Y.; Wang, Z.; Ren, Y.; Lin, T. Metabolomics Response for Drought Stress Tolerance in Chinese Wheat Genotypes (Triticum aestivum). Plants 2020, 9, 520. [Google Scholar] [CrossRef] [Green Version]
- Buffagni, V.; Vurro, F.; Janni, M.; Gullì, M.; Keller, A.A.; Marmiroli, N. Shaping durum wheat for the future: Gene expression analyses and metabolites profiling support the contribution of BCAT genes to drought stress response. Front. Plant Sci. 2020, 11, 891. [Google Scholar] [CrossRef]
- Hong, Y.; Ni, S.-J.; Zhang, G.-P. Transcriptome and metabolome analysis reveals regulatory networks and key genes controlling barley malting quality in responses to drought stress. Plant Physiol. Biochem. 2020, 152, 1–11. [Google Scholar] [CrossRef]
- Cao, L.; Jin, X.; Zhang, Y.; Zhang, M.; Wang, Y. Transcriptomic and metabolomic profiling of melatonin treated soybean (Glycine max L.) under drought stress during grain filling period through regulation of secondary metabolite biosynthesis pathways. PLoS ONE 2020, 15, e0239701. [Google Scholar] [CrossRef]
- Han, Z.; Ahsan, M.; Adil, M.F.; Chen, X.; Nazir, M.M.; Shamsi, I.H.; Zeng, F.; Zhang, G. Identification of the gene network modules highly associated with the synthesis of phenolics compounds in barley by transcriptome and metabolome analysis. Food Chem. 2020, 323, 126862. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, X.; Lu, X.; Zhao, B.; Yang, Y.; Liu, J. Integrative analysis of transcriptome and metabolome reveal mechanism of tolerance to salt stress in oat (Avena sativa L.). Plant Physiol. Biochem. 2021, 160, 315–328. [Google Scholar] [CrossRef]
- Wang, W.; Pang, J.; Zhang, F.; Sun, L.; Yang, L.; Zhao, Y.; Yang, Y.; Wang, Y.; Siddique, K.H. Integrated transcriptomics and metabolomics analysis to characterize alkali stress responses in canola (Brassica napus L.). Plant Physiol. Biochem. 2021, 166, 605–620. [Google Scholar] [CrossRef]
- Pan, J.; Li, Z.; Dai, S.; Ding, H.; Wang, Q.; Li, X.; Ding, G.; Wang, P.; Guan, Y.; Liu, W. Integrative analyses of transcriptomics and metabolomics upon seed germination of foxtail millet in response to salinity. Sci. Rep. 2020, 10, 13660. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Kim, J.K.; Jia, C.; Yin, F.; Kim, H.J.; Akram, W.; Hu, X.; Li, X. Comparative transcriptome and metabolic profiling analysis of buckwheat (Fagopyrum tataricum (L.) Gaertn.) under salinity stress. Metabolites 2019, 9, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wang, B.; Liu, D.; Zou, C.; Wu, P.; Wang, Z.; Wang, Y.; Li, C. Transcriptomic and metabolomic analyses reveal mechanisms of adaptation to salinity in which carbon and nitrogen metabolism is altered in sugar beet roots. BMC Plant Biol. 2020, 20, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Kong, L.; Zhang, Y.; Liao, Y. Gene and Metabolite Integration Analysis through Transcriptome and Metabolome Brings New Insight into Heat Stress Tolerance in Potato (Solanum tuberosum L.). Plants 2021, 10, 103. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, M.; Xu, K.; Li, J.; Li, S.; Zhang, S.; Yang, X. Integrated transcriptomics and metabolomics analyses provide insights into cold stress response in wheat. Crop J. 2019, 7, 857–866. [Google Scholar] [CrossRef]
- Gupta, A.; Patil, M.; Qamar, A.; Senthil-Kumar, M. ath-miR164c influences plant responses to the combined stress of drought and bacterial infection by regulating proline metabolism. Environ. Exp. Bot. 2020, 172, 103998. [Google Scholar] [CrossRef]
- Muthuramalingam, P.; Jeyasri, R.; Selvaraj, A.; Pandian, S.K.; Ramesh, M. Integrated transcriptomic and metabolomic analyses of glutamine metabolism genes unveil key players in Oryza sativa (L.) to ameliorate the unique and combined abiotic stress tolerance. Int. J. Biol. Macromol. 2020, 164, 222–231. [Google Scholar] [CrossRef]
- Redenbaugh, K.; Hiatt, W.; Martineau, B.; Emlay, D. Regulatory assessment of the FLAVR SAVR tomato. Trends Food Sci. Technol. 1994, 5, 105–110. [Google Scholar] [CrossRef]
- Padgette, S.R.; Kolacz, K.H.; Delannay, X.; Re, D.; LaVallee, B.; Tinius, C.; Rhodes, W.; Otero, Y.; Barry, G.; Eichholtz, D. Development, identification, and characterization of a glyphosate-tolerant soybean line. Crop Sci. 1995, 35, 1451–1461. [Google Scholar] [CrossRef]
- Razzaq, A.; Saleem, F.; Kanwal, M.; Mustafa, G.; Yousaf, S.; Imran Arshad, H.M.; Hameed, M.K.; Khan, M.S.; Joyia, F.A. Modern trends in plant genome editing: An inclusive review of the CRISPR/Cas9 toolbox. Int. J. Mol. Sci. 2019, 20, 4045. [Google Scholar] [CrossRef] [Green Version]
- Waltz, E. CRISPR-edited crops free to enter market, skip regulation. Nat. Biotechnol. 2016, 34, 582–583. [Google Scholar] [CrossRef] [PubMed]
- Tatsis, E.C.; O’Connor, S.E. New developments in engineering plant metabolic pathways. Curr. Opin. Biotechnol. 2016, 42, 126–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkina, L. Phenylpropanoids as naturally occurring antioxidants: From plant defense to human health. Cell Mol. Biol. 2007, 53, 15–25. [Google Scholar] [PubMed]
- Jamil, I.N.; Remali, J.; Azizan, K.A.; Nor Muhammad, N.A.; Arita, M.; Goh, H.-H.; Aizat, W.M. Systematic multi-omics integration (MOI) approach in plant systems biology. Front. Plant Sci. 2020, 11, 944. [Google Scholar] [CrossRef] [PubMed]
- Ganjewala, D.; Kaur, G.; Srivastava, N. Metabolic engineering of stress protectant secondary metabolites to confer abiotic stress tolerance in plants. In Molecular Approaches in Plant Biology and Environmental Challenges; Springer: Singapore, 2019; pp. 207–227. [Google Scholar]
- Kumar, V.; Baweja, M.; Singh, P.K.; Shukla, P. Recent developments in systems biology and metabolic engineering of plant–microbe interactions. Front. Plant Sci. 2016, 7, 1421. [Google Scholar] [CrossRef]
- Yang, T.; Stoopen, G.; Yalpani, N.; Vervoort, J.; de Vos, R.; Voster, A.; Verstappen, F.W.; Bouwmeester, H.J.; Jongsma, M.A. Metabolic engineering of geranic acid in maize to achieve fungal resistance is compromised by novel glycosylation patterns. Metab. Eng. 2011, 13, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Blancquaert, D.; De Steur, H.; Gellynck, X.; Van Der Straeten, D. Metabolic engineering of micronutrients in crop plants. Ann. N. Y. Acad. Sci. 2017, 1390, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.W.; Kim, J.; Kwon, S.I.; Corvalán, C.; Cho, S.W.; Kim, H.; Kim, S.-G.; Kim, S.-T.; Choe, S.; Kim, J.-S. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat. Biotechnol. 2015, 33, 1162–1164. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, J.-K. Precise editing of a target base in the rice genome using a modified CRISPR/Cas9 system. Mol. Plant 2017, 10, 523–525. [Google Scholar] [CrossRef] [Green Version]
- Schouten, H.J.; Krens, F.A.; Jacobsen, E. Cisgenic plants are similar to traditionally bred plants: International regulations for genetically modified organisms should be altered to exempt cisgenesis. EMBO Rep. 2006, 7, 750–753. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Li, R.; Li, X.; Fu, D.; Zhu, B.; Tian, H.; Luo, Y.; Zhu, H. Multiplexed CRISPR/Cas9-mediated metabolic engineering of γ-aminobutyric acid levels in Solanum lycopersicum. Plant Biotechnol. J. 2018, 16, 415–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najera, V.A.; Twyman, R.M.; Christou, P.; Zhu, C. Applications of multiplex genome editing in higher plants. Curr. Opin. Biotechnol. 2019, 59, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Alagoz, Y.; Gurkok, T.; Zhang, B.; Unver, T. Manipulating the biosynthesis of bioactive compound alkaloids for next-generation metabolic engineering in opium poppy using CRISPR-Cas 9 genome editing technology. Sci. Rep. 2016, 6, 30910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, Y.; Chen, S.; Tian, H.; Fu, D.; Zhu, B.; Luo, Y.; Zhu, H. Lycopene is enriched in tomato fruit by CRISPR/Cas9-mediated multiplex genome editing. Front. Plant Sci. 2018, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Du, H.; Wang, J.; Pu, Y.; Yang, C.; Yan, R.; Yang, H.; Cheng, H.; Yu, D. Multiplex CRISPR/Cas9-mediated metabolic engineering increases soya bean isoflavone content and resistance to soya bean mosaic virus. Plant Biotechnol. J. 2020, 18, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, G.; Goossens, A.; Colinas, M. Metabolic editing: Small measures, great impact. Curr. Opin. Biotechnol. 2019, 59, 16–23. [Google Scholar] [CrossRef]
- Schindel, H.S.; Piatek, A.A.; Stewart, C.N.; Lenaghan, S.C. The plastid genome as a chassis for synthetic biology-enabled metabolic engineering: Players in gene expression. Plant Cell Rep. 2018, 37, 1419–1429. [Google Scholar] [CrossRef]
- Farre, G.; Twyman, R.M.; Christou, P.; Capell, T.; Zhu, C. Knowledge-driven approaches for engineering complex metabolic pathways in plants. Curr. Opin. Biotechnol. 2015, 32, 54–60. [Google Scholar] [CrossRef]
- Christ, B.; Pluskal, T.; Aubry, S.; Weng, J.-K. Contribution of untargeted metabolomics for future assessment of biotech crops. Trends Plant Sci. 2018, 23, 1047–1056. [Google Scholar] [CrossRef]
- Baker, J.M.; Hawkins, N.D.; Ward, J.L.; Lovegrove, A.; Napier, J.A.; Shewry, P.R.; Beale, M.H. A metabolomic study of substantial equivalence of field-grown genetically modified wheat. Plant Biotechnol. J. 2006, 4, 381–392. [Google Scholar] [CrossRef]
- Kogel, K.-H.; Voll, L.M.; Schäfer, P.; Jansen, C.; Wu, Y.; Langen, G.; Imani, J.; Hofmann, J.; Schmiedl, A.; Sonnewald, S. Transcriptome and metabolome profiling of field-grown transgenic barley lack induced differences but show cultivar-specific variances. Proc. Natl. Acad. Sci. USA 2010, 107, 6198–6203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, L.V.T.; Hackett, C.A.; Alexander, C.J.; McNicol, J.W.; Sungurtas, J.A.; Stewart, D.; McCue, K.F.; Belknap, W.R.; Davies, H.V. Modifying glycoalkaloid content in transgenic potato–Metabolome impacts. Food Chem. 2015, 187, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Bernillon, S.; Maucourt, M.; Deborde, C.; Chéreau, S.; Jacob, D.; Priymenko, N.; Laporte, B.; Coumoul, X.; Salles, B.; Rogowsky, P.M. Characterization of GMO or glyphosate effects on the composition of maize grain and maize-based diet for rat feeding. Metabolomics 2018, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Yang, L.; Guo, J.; Quan, S.; Chen, G.; Zhao, X.; Zhang, D.; Shi, J. Metabolic changes in transgenic maize mature seeds over-expressing the Aspergillus niger phyA2. Plant Cell Rep. 2016, 35, 429–437. [Google Scholar] [CrossRef]
- Clarke, J.D.; Alexander, D.C.; Ward, D.P.; Ryals, J.A.; Mitchell, M.W.; Wulff, J.E.; Guo, L. Assessment of genetically modified soybean in relation to natural variation in the soybean seed metabolome. Sci. Rep. 2013, 3, 3082. [Google Scholar] [CrossRef]
- Catchpole, G.S.; Beckmann, M.; Enot, D.P.; Mondhe, M.; Zywicki, B.; Taylor, J.; Hardy, N.; Smith, A.; King, R.D.; Kell, D.B. Hierarchical metabolomics demonstrates substantial compositional similarity between genetically modified and conventional potato crops. Proc. Natl. Acad. Sci. USA 2005, 102, 14458–14462. [Google Scholar] [CrossRef] [Green Version]
- Kusano, M.; Redestig, H.; Hirai, T.; Oikawa, A.; Matsuda, F.; Fukushima, A.; Arita, M.; Watanabe, S.; Yano, M.; Hiwasa-Tanase, K. Covering chemical diversity of genetically-modified tomatoes using metabolomics for objective substantial equivalence assessment. PLoS ONE 2011, 6, e16989. [Google Scholar] [CrossRef] [Green Version]
- Kusano, M.; Baxter, I.; Fukushima, A.; Oikawa, A.; Okazaki, Y.; Nakabayashi, R.; Bouvrette, D.J.; Achard, F.; Jakubowski, A.R.; Ballam, J.M. Assessing metabolomic and chemical diversity of a soybean lineage representing 35 years of breeding. Metabolomics 2015, 11, 261–270. [Google Scholar] [CrossRef]
- Fraser, P.D.; Aharoni, A.; Hall, R.D.; Huang, S.; Giovannoni, J.J.; Sonnewald, U.; Fernie, A.R. Metabolomics should be deployed in the identification and characterization of gene-edited crops. Plant J. 2020, 102, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.; Ghosh, S.; Williams, M.J.; Cuddy, W.S.; Simmonds, J.; Rey, M.-D.; Hatta, M.A.M.; Hinchliffe, A.; Steed, A.; Reynolds, D. Speed breeding is a powerful tool to accelerate crop research and breeding. Nat. Plants 2018, 4, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Watson, A.; Gonzalez-Navarro, O.E.; Ramirez-Gonzalez, R.H.; Yanes, L.; Mendoza-Suárez, M.; Simmonds, J.; Wells, R.; Rayner, T.; Green, P. Speed breeding in growth chambers and glasshouses for crop breeding and model plant research. Nat. Protoc. 2018, 13, 2944–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jähne, F.; Hahn, V.; Würschum, T.; Leiser, W.L. Speed breeding short-day crops by LED-controlled light schemes. Theor. Appl. Genet. 2020, 80, 100. [Google Scholar] [CrossRef] [PubMed]
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crop | Analytical Tool | Detected Metabolites | No. of Candidate Genes | No. of QTLs | Trait Study | Reference | |
|---|---|---|---|---|---|---|---|
| Rice | mGWAS | LC-MS/MS | l-alanine, l-tyramine, threonine, leucine and histidine Syringenone, Chlorogenic acid | 36 | 356 | Nutritional value | [26] |
| Peral Millet | FIE-HRMS, | vitamins, antioxidants, dietary starch | 738 | 987 | Nutritional improvement | [27] | |
| Rice | LC-MS/MS | Amino acids, flavonoids | 58 | 24 | Grain color, size, and weight | [28] | |
| Wheat | GC-MS | L-tyrosine, pentose alcohol III, L-arginine, ornithine, oxalic acid, | 25 | 38 | Association between metabolic phenotypes | [29] | |
| Maize | LC-MS/MS | Flavonoid, benzoxazinoid | - | - | Pathogen resistance | [30] | |
| Maize | LC-MS | Terpenoids, benzoxazinoids, lipids, amino acids, flavonoids, | 10 | - | Salt tolerance | [31] | |
| Soybean | LC-MS | Alanine, arginine, asparagine, aspartic acid, daidzein | 284 | 144 | Seed oil-related traits | [32] | |
| Foxtail Millet | LC-ESI-MS/MS | lipids, hydroxycinnamoyl derivatives, phenolamides and flavonoids | 5 | 237 | Environment adaptation | [33] | |
| Wheat | LC-MS/MS | Flavonoids | 26 | 42 | Flavonoid pathways | [34] | |
| Tomato | ESI-QqTOF-MS/MS | Amino acid, alkaloids, vitamins, polyamine, polyphenol | 535 | - | Fruit traits | [35] | |
| Wheat | LC-MS/MS | Betaine, deoxyinosine-5′-monophosphate | 24 | 1005 | Grains per spike, plant height | [36] | |
| Barley | LC-MS | Glycosides, acylated glycosides of flavones, phenylpropenoic acid | - | 138 | Drought tolerance | [37] | |
| Barley | MS (IC-MS/MS) | succinate, glutathione, γ-tocopherol | - | 13 | Drought and heat stress | [38] | |
| Tomato | GC/MS, UPLC | Acyl-sugars, glycoalkaloids, flavonols | - | 212 | Fruit metabolism | [39] | |
| Tomato | UPLC | Glycoalkaloids, acyl-sugar, hydroxycinnamates | 4 | 679 | Environmental stress tolerance | [40] | |
| Strawberry | LC-ESI-MS | Phenolics, flavonoids, anthocyanins | - | 178 | Fruit quality | [41] | |
| Rice | LC-MS/MS | L-asparagine, feruloylserotonin | 35 | 4681 | Agronomic traits | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razzaq, A.; Wishart, D.S.; Wani, S.H.; Hameed, M.K.; Mubin, M.; Saleem, F. Advances in Metabolomics-Driven Diagnostic Breeding and Crop Improvement. Metabolites 2022, 12, 511. https://doi.org/10.3390/metabo12060511
Razzaq A, Wishart DS, Wani SH, Hameed MK, Mubin M, Saleem F. Advances in Metabolomics-Driven Diagnostic Breeding and Crop Improvement. Metabolites. 2022; 12(6):511. https://doi.org/10.3390/metabo12060511
Chicago/Turabian StyleRazzaq, Ali, David S. Wishart, Shabir Hussain Wani, Muhammad Khalid Hameed, Muhammad Mubin, and Fozia Saleem. 2022. "Advances in Metabolomics-Driven Diagnostic Breeding and Crop Improvement" Metabolites 12, no. 6: 511. https://doi.org/10.3390/metabo12060511
APA StyleRazzaq, A., Wishart, D. S., Wani, S. H., Hameed, M. K., Mubin, M., & Saleem, F. (2022). Advances in Metabolomics-Driven Diagnostic Breeding and Crop Improvement. Metabolites, 12(6), 511. https://doi.org/10.3390/metabo12060511