
The Potential Role of Metabolomics in Drug-Induced Liver Injury (DILI) Assessment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. DILI and Its Clinical and Pharmaceutical Relevance
1.2. Types of DILI and Clinical Features of DILI
1.3. The Diagnosis of DILI: Conventional Clinical and Biochemical Biomarkers
2. Metabolomics in DILI Research and Diagnosis
2.1. Metabolomics vs. Transcriptomics and Proteomics in DILI Biomarker Discovery
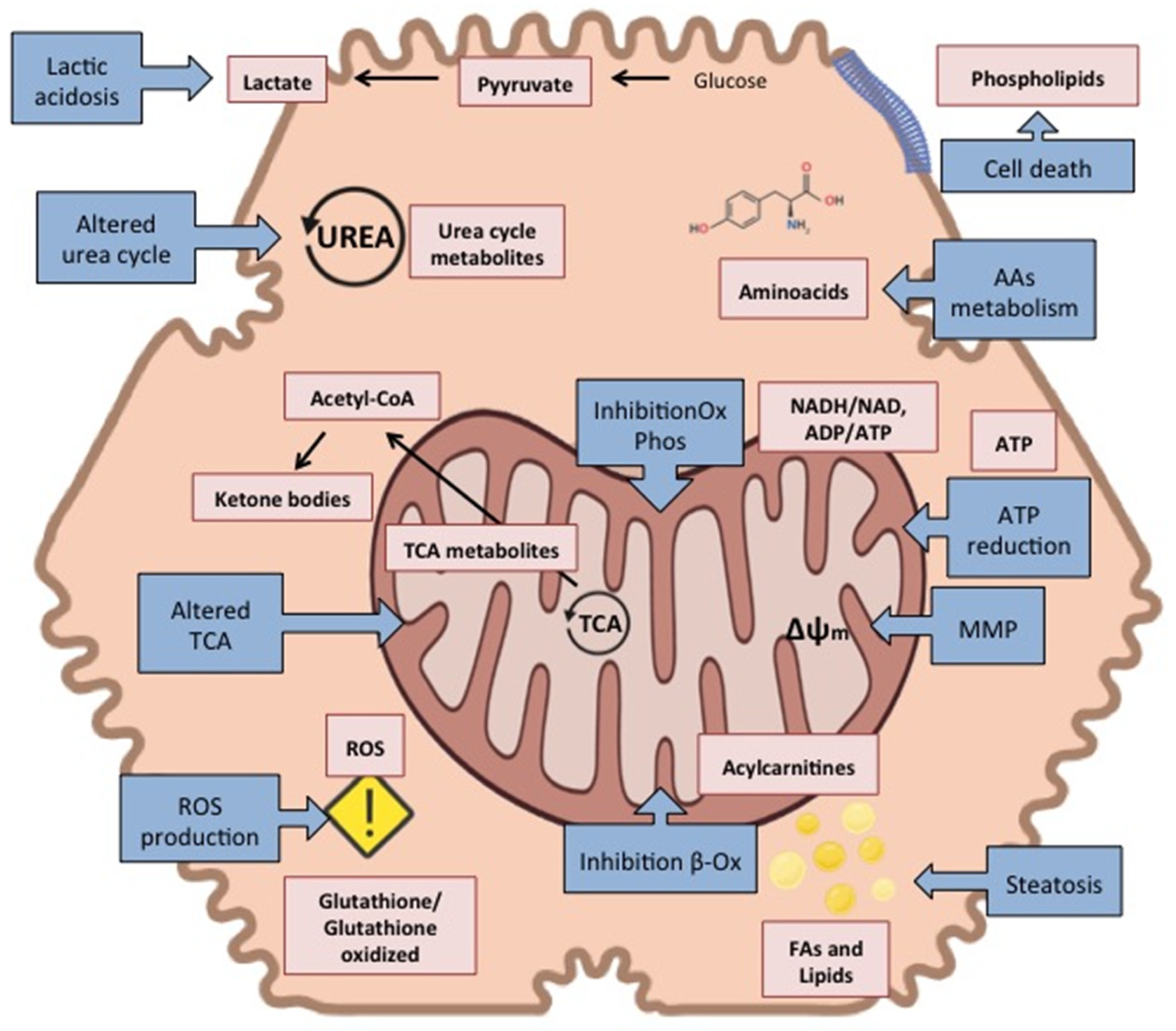
2.2. Metabolomics for the Study of DILI Pathophysiology
2.3. General Principles for MS-Based Metabolomics Analysis and Biomarker Identification in DILI
2.4. Metabolites Identified as Putative Biomarkers of DILI in Humans
3. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vuppalanchi, R.; Liangpunsakul, S.; Chalasani, N. Etiology of new-onset jaundice: How often is it caused by idiosyncratic drug-induced liver injury in the United States? Am. J. Gastroenterol. 2007, 102, 558–693; quiz 693. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.A.; Ryder, S.; Lavado, A.; Dilworth, C.; Riley, R.J. The evolution of strategies to minimise the risk of human drug-induced liver injury (DILI) in drug discovery and development. Arch. Toxicol. 2020, 94, 2559–2585. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.; Reisch, J.S.; Schiødt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Ostapowicz, G.; Fontana, R.J.; Schiødt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a Prospective Study of Acute Liver Failure at 17 Tertiary Care Centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef]
- Björnsson, E.; Jerlstad, P.; Bergqvist, A.; Olsson, R. Fulminant drug-induced hepatic failure leading to death or liver transplantation in Sweden. Scand. J. Gastroenterol. 2005, 40, 1095–1101. [Google Scholar] [CrossRef]
- Russo, M.W.; Galanko, J.A.; Shrestha, R.; Fried, M.W.; Watkins, P. Liver transplantation for acute liver failure from drug induced liver injury in the United States. Liver Transplant. 2004, 10, 1018–1023. [Google Scholar] [CrossRef]
- Björnsson, E.S.; Bergmann, O.M.; Björnsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, Presentation, and Outcomes in Patients with Drug-Induced Liver Injury in the General Population of Iceland. Gastroenterology 2013, 144, 1419–1425.e3. [Google Scholar] [CrossRef]
- Saxena, V.; Gupta, A.; Gowda, G.A.N.; Saxena, R.; Yachha, S.K.; Khetrapal, C.L. 1H NMR spectroscopy for the prediction of therapeutic outcome in patients with fulminant hepatic failure. NMR Biomed. 2006, 19, 521–526. [Google Scholar] [CrossRef]
- De Abajo, F.J.; Montero, D.; Madurga, M.; Rodríguez, L.A.G. Acute and clinically relevant drug-induced liver injury: A population based case-control study. Br. J. Clin. Pharmacol. 2004, 58, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Regev, A. Drug-Induced Liver Injury and Drug Development: Industry Perspective. Semin. Liver Dis. 2014, 34, 227–239. [Google Scholar] [CrossRef]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-induced liver injury: Interactions between drug properties and host factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Castell, J.V. Allergic hepatitis: A drug-mediated organ-specific immune reaction. Clin. Exp. Allergy 1998, 28 (Suppl. S4), 13–19. [Google Scholar] [PubMed]
- Castell, J.V.; Castell, M. Allergic hepatitis induced by drugs. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-C.; Mao, Y.-M.; Chen, C.-W.; Chen, J.-J.; Chen, J.; Cong, W.-M.; Ding, Y.; Duan, Z.-P.; Fu, Q.-C.; Guo, X.-Y.; et al. CSH guidelines for the diagnosis and treatment of drug-induced liver injury. Hepatol. Int. 2017, 11, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, N.; Navarro, V. Drug-Induced Liver Injury in GI Practice. Hepatol. Commun. 2020, 4, 631–645. [Google Scholar] [CrossRef]
- Kleiner, D.E. The Pathology of Drug-Induced Liver Injury. Semin. Liver Dis. 2009, 29, 364–372. [Google Scholar] [CrossRef]
- Tillmann, H.L.; Suzuki, A.; Barnhart, H.X.; Serrano, J.; Rockey, D.C. Tools for causality assessment in drug-induced liver disease. Curr. Opin. Gastroenterol. 2019, 35, 183–190. [Google Scholar] [CrossRef]
- Lucena, M.I.; Andrade, R.J.; Fernández, M.C.; Pachkoria, K.; Pelaez, G.; Durán, J.A.; Villar, M.; Rodrigo, L.; Romero-Gomez, M.; Planas, R.; et al. Determinants of the clinical expression of amoxicillin-clavulanate hepatotoxicity: A prospective series from Spain. Hepatology 2006, 44, 850–856. [Google Scholar] [CrossRef]
- Lucena, M.I.; Andrade, R.J.; Rodrigo, L.; Salmerón, J.; Alvarez, A.; Lopez-Garrido, M.J.; Camargo, R.; Alcantára, R. Trovafloxacin-Induced Acute Hepatitis. Clin. Infect. Dis. 2000, 30, 400–401. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.M.; Senior, J.R. Recognizing Drug-Induced Liver Injury: Current Problems, Possible Solutions. Toxicol. Pathol. 2005, 33, 155–164. [Google Scholar] [CrossRef]
- Danjuma, M.I.M.U.; Sajid, J.; Fatima, H.; Elzouki, A.N. Novel biomarkers for potential risk stratification of drug induced liver injury (DILI): A narrative perspective on current trends. Medicine 2019, 98, e18322. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Schnackenberg, L.K.; Shi, Q.; Salminen, W.F. Chapter 13—Hepatic toxicity biomarkers. In Biomarkers in Toxicology; Academic Press: Cambridge, MA, USA, 2014; pp. 241–259. [Google Scholar]
- Watkins, P.B. Managing the risk of drug-induced liver injury. Clin. Pharmacol. Ther. 2013, 94, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J.; Hayashi, P.H.; Gu, J.; Reddy, K.R.; Barnhart, H.; Watkins, P.B.; Serrano, J.; Lee, W.M.; Chalasani, N.; Stolz, A.; et al. Idiosyncratic Drug-Induced Liver Injury Is Associated with Substantial Morbidity and Mortality within 6 Months from Onset. Gastroenterology 2014, 147, 96–108.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Robinson, S.D. Drug-Induced Liver Disease; Kaplowitz, N., DeLeve, L.D., Eds.; Marcel Decker: New York, NY, USA, 2003; p. 773. ISBN 0-8247-0811-3. [Google Scholar]
- Devarbhavi, H. An Update on Drug-induced Liver Injury. J. Clin. Exp. Hepatol. 2012, 2, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Amacher, D.E.; Schomaker, S.J.; Aubrecht, J. Development of Blood Biomarkers for Drug-Induced Liver Injury: An Evaluation of Their Potential for Risk Assessment and Diagnostics. Mol. Diagn. Ther. 2013, 17, 343–354. [Google Scholar] [CrossRef]
- Russmann, S.; Kullak-Ublick, G.A.; Grattagliano, I. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr. Med. Chem. 2009, 16, 3041–3053. [Google Scholar] [CrossRef] [Green Version]
- Antoine, D.J.; Dear, J.W.; Lewis, P.S.; Platt, V.; Coyle, J.; Masson, M.; Thanacoody, R.H.; Gray, A.J.; Webb, D.J.; Moggs, J.G.; et al. Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital. Hepatology 2013, 58, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Church, R.J.; Kullak-Ublick, G.A.; Aubrecht, J.; Bonkovsky, H.L.; Chalasani, N.; Fontana, R.J.; Goepfert, J.C.; Hackman, F.; King, N.M.P.; Kirby, S.; et al. Candidate biomarkers for the diagnosis and prognosis of drug-induced liver injury: An international collaborative effort. Hepatology 2019, 69, 760–773. [Google Scholar] [CrossRef]
- Fu, S.; Wu, D.; Jiang, W.; Li, J.; Long, J.; Jia, C.; Zhou, T. Molecular Biomarkers in Drug-Induced Liver Injury: Challenges and Future Perspectives. Front. Pharmacol. 2020, 10, 1667. [Google Scholar] [CrossRef] [Green Version]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef]
- Singer, J.B.; Lewitzky, S.; Leroy, E.; Yang, F.; Zhao, X.; Klickstein, L.; Wright, T.M.; Meyer, J.; Paulding, C.A. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat. Genet. 2010, 42, 711–714. [Google Scholar] [CrossRef]
- Mallal, S.; Phillips, E.; Carosi, G.; Molina, J.-M.; Workman, C.; Tomažič, J.; Jägel-Guedes, E.; Rugina, S.; Kozyrev, O.; Cid, J.F.; et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. N. Engl. J. Med. 2008, 358, 568–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, N.J.; Waterfield, C.J.; Farrant, R.D.; Holmes, A.E.; Nicholson, J.K. Integrated Metabonomic Analysis of Bromobenzene-Induced Hepatotoxicity: Novel Induction of 5-Oxoprolinosis. J. Proteome Res. 2006, 5, 1448–1459. [Google Scholar] [CrossRef]
- Vazquez, J.H.; McGill, M.R. Redrawing the Map to Novel DILI Biomarkers in Circulation: Where Are We, Where Should We Go, and How Can We Get There? Livers 2021, 1, 22. [Google Scholar] [CrossRef]
- Abraham, V.C.; Towne, D.L.; Waring, J.F.; Warrior, U.; Burns, D.J. Application of a High-Content Multiparameter Cytotoxicity Assay to Prioritize Compounds Based on Toxicity Potential in Humans. SLAS Discov. Adv. Sci. Drug Discov. 2008, 13, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Benet, M.; Moya, M.; Donato, M.T.; Lahoz, A.; Hervás, D.; Guzmán, C.; Gómez-Lechón, M.J.; Castell, J.V.; Jover, R. A simple transcriptomic signature able to predict drug-induced hepatic steatosis. Arch. Toxicol. 2014, 88, 967–982. [Google Scholar] [CrossRef] [PubMed]
- Bleibel, W.; Kim, S.; D’Silva, K.; Lemmer, E.R. Drug-Induced Liver Injury: Review Article. Am. J. Dig. Dis. 2007, 52, 2463–2471. [Google Scholar] [CrossRef]
- Iruzubieta, P.; Arias-Loste, M.T.; Barbier-Torres, L.; Martinez-Chantar, M.L.; Crespo, J. The Need for Biomarkers in Diagnosis and Prognosis of Drug-Induced Liver Disease: Does Metabolomics Have Any Role? BioMed Res. Int. 2015, 2015, 386186. [Google Scholar] [CrossRef] [Green Version]
- Oldiges, M.; Lütz, S.; Pflug, S.; Schroer, K.; Stein, N.; Wiendahl, C. Metabolomics: Current state and evolving methodologies and tools. Appl. Microbiol. Biotechnol. 2007, 76, 495–511. [Google Scholar] [CrossRef]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef]
- Clayton, T.A.; Baker, D.; Lindon, J.C.; Everett, J.R.; Nicholson, J.K. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 14728–14733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Huang, R.; Liu, L.; Peng, J.; Xiao, B.; Yang, J.; Miao, Z.; Huang, H. Metabonomics study of urine from Sprague–Dawley rats exposed to Huang-yao-zi using 1H NMR spectroscopy. J. Pharm. Biomed. Anal. 2010, 52, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wang, Y.; Sheng, Z.; Liu, G.; Fu, Z.; Zhao, J.; Zhao, J.; Yan, X.; Zhu, B.; Peng, S. NMR-based metabonomic analysis of the hepatotoxicity induced by combined exposure to PCBs and TCDD in rats. Toxicol. Appl. Pharmacol. 2010, 248, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Miyawaki, I.; Bando, K.; Horie, H.; Zaitsu, K.; Katagi, M.; Bamba, T.; Tsuchihashi, H.; Fukusaki, E. Influences of methamphetamine-induced acute intoxication on urinary and plasma metabolic profiles in the rat. Toxicology 2011, 287, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Winnike, J.H.; Li, Z.; Wright, F.A.; Macdonald, J.M.; O’Connell, T.M.; Watkins, P.B. Use of Pharmaco-Metabonomics for Early Prediction of Acetaminophen-Induced Hepatotoxicity in Humans. Clin. Pharmacol. Ther. 2010, 88, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Xie, Y.; McGill, M. Acetaminophen-induced Liver Injury: From Animal Models to Humans. J. Clin. Transl. Hepatol. 2014, 2, 153–161. [Google Scholar] [CrossRef]
- García-Cañaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J. Lipid Res. 2012, 53, 2231–2241. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, V.; Björnsson, E.S. Drug-induced cholestasis. Hepatol. Commun. 2017, 1, 726–735. [Google Scholar] [CrossRef]
- Kawada, N.; Kristensen, D.B.; Asahina, K.; Nakatani, K.; Minamiyama, Y.; Seki, S.; Yoshizato, K. Characterization of a stellate cell activation-associated protein (STAP) with peroxidase activity found in rat hepatic stellate cells. J. Biol. Chem. 2001, 276, 25318–25323. [Google Scholar] [CrossRef] [Green Version]
- Teranishi, Y.; Matsubara, T.; Krausz, K.W.; Le, T.T.T.; Gonzalez, F.J.; Yoshizato, K.; Ikeda, K.; Kawada, N. Involvement of hepatic stellate cell cytoglobin in acute hepatocyte damage through the regulation of CYP2E1-mediated xenobiotic metabolism. Lab. Investig. 2015, 95, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Lecoeur, M.; Rabenirina, G.; Schifano, N.; Odou, P.; Ethgen, S.; Lebuffe, G.; Foulon, C. Determination of acetaminophen and its main metabolites in urine by capillary electrophoresis hyphenated to mass spectrometry. Talanta 2019, 205, 120108. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Torres, M.; García-Llorens, G.; Moro, E.; Méndez, R.; Quintás, G.; Castell, J.V. Factors that influence the quality of metabolomics data in in vitro cell toxicity studies: A systematic survey. Sci. Rep. 2021, 11, 22119. [Google Scholar] [CrossRef] [PubMed]
- Fannin, R.D.; Russo, M.; O’Connell, T.M.; Gerrish, K.; Winnike, J.H.; Macdonald, J.; Newton, J.; Malik, S.; Sieber, S.O.; Parker, J.; et al. Acetaminophen dosing of humans results in blood transcriptome and metabolome changes consistent with impaired oxidative phosphorylation. Hepatology 2009, 51, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Sun, J.; Schnackenberg, L.K. Metabolomics approaches for discovering biomarkers of drug-induced hepatotoxicity and nephrotoxicity. Toxicol. Appl. Pharmacol. 2010, 243, 154–166. [Google Scholar] [CrossRef]
- Yu, M.; Zhu, Y.; Cong, Q.; Wu, C. Metabonomics Research Progress on Liver Diseases. Can. J. Gastroenterol. Hepatol. 2017, 2017, 8467192. [Google Scholar] [CrossRef]
- Beckonert, O.; Coen, M.; Keun, H.C.; Wang, Y.; Ebbels, T.M.D.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. High-resolution magic-angle-spinning NMR spectroscopy for metabolic profiling of intact tissues. Nat. Protoc. 2010, 5, 1019–1032. [Google Scholar] [CrossRef]
- Zimmerman, H.J. Drug-induced liver disease. Clin. Liver Dis. 2000, 4, 73–96. [Google Scholar] [CrossRef]
- Lenz, E.M.; Wilson, I.D. Analytical Strategies in Metabonomics. J. Proteome Res. 2006, 6, 443–458. [Google Scholar] [CrossRef]
- Beger, R.D. A Review of Applications of Metabolomics in Cancer. Metabolites 2013, 3, 552–574. [Google Scholar] [CrossRef] [Green Version]
- Robards, K.; Haddad, P.; Jackson, P. Principles and Practice of Modern Chromatographic Methods; Academic Press: Cambridge, MA, USA, 1994. [Google Scholar] [CrossRef]
- Misra, B.B.; Bassey, E.; Olivier, M. Comparison of a GC-Orbitrap-MS with parallel GC-FID capabilities for metabolomics of human serum. bioRxiv 2019, 740795. [Google Scholar] [CrossRef] [Green Version]
- Hawrył, A.; Hawrył, M.; Waksmundzka-Hajnos, M. Liquid chromatography fingerprint analysis and antioxidant activity of selected lavender species with chemometric calculations. PLoS ONE 2019, 14, e0218974. [Google Scholar] [CrossRef]
- Zheng, Q.-X.; Fu, H.-Y.; Li, H.-D.; Wang, B.; Peng, C.-H.; Wang, S.; Cai, J.-L.; Liu, S.-F.; Zhang, X.-B.; Yu, Y.-J. Automatic time-shift alignment method for chromatographic data analysis. Sci. Rep. 2017, 7, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Monge, M.E.; Dodds, J.N.; Baker, E.S.; Edison, A.S.; Fernández, F.M. Challenges in Identifying the Dark Molecules of Life. Annu. Rev. Anal. Chem. 2019, 12, 177–199. [Google Scholar] [CrossRef]
- Ten-Doménech, I.; Martínez-Sena, T.; Moreno-Torres, M.; Sanjuan-Herráez, J.D.; Castell, J.V.; Parra-Llorca, A.; Vento, M.; Quintás, G.; Kuligowski, J. Comparing Targeted vs. Untargeted MS2 Data-Dependent Acquisition for Peak Annotation in LC–MS Metabolomics. Metabolites 2020, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Zelena, E.; Dunn, W.B.; Broadhurst, D.; Francis-McIntyre, S.; Carroll, K.M.; Begley, P.; O’Hagan, S.; Knowles, J.D.; Halsall, A.; Wilson, I.D.; et al. Development of a Robust and Repeatable UPLC−MS Method for the Long-Term Metabolomic Study of Human Serum. Anal. Chem. 2009, 81, 1357–1364. [Google Scholar] [CrossRef]
- Sánchez-Illana, Á.; Perez-Guaita, D.; Cuesta-García, D.; Sanjuan-Herráez, J.D.; Vento, M.; Ruiz-Cerdá, J.L.; Quintás, G.; Kuligowski, J. Model selection for within-batch effect correction in UPLC-MS metabolomics using quality control—Support vector regression. Anal. Chim. Acta 2018, 1026, 62–68. [Google Scholar] [CrossRef]
- Sánchez-Illana, Á.; Piñeiro-Ramos, J.D.; Sanjuan-Herráez, J.D.; Vento, M.; Quintás, G.; Kuligowski, J. Evaluation of batch effect elimination using quality control replicates in LC-MS metabolite profiling. Anal. Chim. Acta 2018, 1019, 38–48. [Google Scholar] [CrossRef]
- Ten-Doménech, I.; Moreno-Torres, M.; Castell, J.V.; Quintás, G.; Kuligowski, J. Extracting consistent biological information from functional results of metabolomic pathway analysis using the Mantel’s test. Anal. Chim. Acta 2021, 1187, 339173. [Google Scholar] [CrossRef]
- Andersson, U.; Lindberg, J.; Wang, S.; Balasubramanian, R.; Marcusson-Ståhl, M.; Hannula, M.; Zeng, C.; Juhasz, P.J.; Kolmert, J.; Bäckström, J.; et al. A systems biology approach to understanding elevated serum alanine transaminase levels in a clinical trial with ximelagatran. Biomarkers 2009, 14, 572–586. [Google Scholar] [CrossRef]
- Zhang, L.; Niu, M.; Wei, A.-W.; Tang, J.-F.; Tu, C.; Bai, Z.-F.; Zou, Z.-S.; Xiao, X.-H.; Liu, Y.-P.; Wang, J.-B. Risk profiling using metabolomic characteristics for susceptible individuals of drug-induced liver injury caused by Polygonum multiflorum. Arch. Toxicol. 2019, 94, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Sonn, B.J.; Heard, K.J.; Heard, S.M.; D’Alessandro, A.; Reynolds, K.M.; Dart, R.C.; Rumack, B.H.; Monte, A.A. Metabolomic markers predictive of hepatic adaptation to therapeutic dosing of acetaminophen. Clin. Toxicol. 2021, 60, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Pence, L.; Beger, R.; Chaudhuri, S.; McCullough, S.; Yan, K.; Simpson, P.; Hennings, L.; Hinson, J.; James, L. Acylcarnitine Profiles in Acetaminophen Toxicity in the Mouse: Comparison to Toxicity, Metabolism and Hepatocyte Regeneration. Metabolites 2013, 3, 606–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, M.R.; Li, F.; Sharpe, M.R.; Williams, C.D.; Curry, S.C.; Ma, X.; Jaeschke, H. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Arch. Toxicol. 2013, 88, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Huo, T.; Chen, X.; Lu, X.; Qu, L.; Liu, Y.; Cai, S. An effective assessment of valproate sodium-induced hepatotoxicity with UPLC–MS and 1HNMR-based metabonomics approach. J. Chromatogr. B 2014, 969, 109–116. [Google Scholar] [CrossRef]
- Woolbright, B.L.; McGill, M.R.; Staggs, V.S.; Winefield, R.D.; Gholami, P.; Olyaee, M.; Sharpe, M.R.; Curry, S.C.; Lee, W.M.; Jaeschke, H.; et al. Glycodeoxycholic Acid Levels as Prognostic Biomarker in Acetaminophen-Induced Acute Liver Failure Patients. Toxicol. Sci. 2014, 142, 436–444. [Google Scholar] [CrossRef] [Green Version]
- James, L.; Yan, K.; Pence, L.; Simpson, P.; Bhattacharyya, S.; Gill, P.; Letzig, L.; Kearns, G.; Beger, R. Comparison of Bile Acids and Acetaminophen Protein Adducts in Children and Adolescents with Acetaminophen Toxicity. PLoS ONE 2015, 10, e0131010. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ma, Z.; Niu, M.; Zhu, Y.; Liang, Q.; Zhao, Y.; Song, J.; Bai, Z.; Zhang, Y.; Zhang, P.; et al. Evidence chain-based causality identification in herb-induced liver injury: Exemplification of a well-known liver-restorative herb Polygonum multiflorum. Front. Med. 2015, 9, 457–467. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Pence, L.; Yan, K.; Gill, P.; Luo, C.; Letzig, L.G.; Simpson, P.M.; Kearns, G.L.; Beger, R.D.; James, L.P. Targeted metabolomic profiling indicates structure-based perturbations in serum phospholipids in children with acetaminophen overdose. Toxicol. Rep. 2016, 3, 747–755. [Google Scholar] [CrossRef]
- Schnackenberg, L.K.; Sun, J.; Bhattacharyya, S.; Gill, P.; James, L.P.; Beger, R.D. Metabolomics Analysis of Urine Samples from Children after Acetaminophen Overdose. Metabolites 2017, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Ji, S.C.; Kim, B.; Yi, S.; Shin, K.H.; Cho, J.Y.; Lim, K.S.; Lee, S.H.; Yoon, S.H.; Chung, J.Y.; et al. Exploration of Biomarkers for Amoxicillin/Clavulanate-Induced Liver Injury: Multi-Omics Approaches. Clin. Transl. Sci. 2016, 10, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Mi, Y.; Shi, C.; Bian, Y.; Huang, C.; Ye, Z.; Liu, L.; Miao, L. First-line anti-tuberculosis drugs induce hepatotoxicity: A novel mechanism based on a urinary metabolomics platform. Biochem. Biophys. Res. Commun. 2018, 497, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, X.; Yin, P.; Wu, R.; Zhou, L.; Xu, G.; Niu, J. Serum metabolome and targeted bile acid profiling reveals potential novel biomarkers for drug-induced liver injury. Medicine 2019, 98, e16717. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Chen, E.; Ouyang, X.; Xu, X.; Ma, S.; Ji, F.; Wu, D.; Zhang, S.; Zhao, Y.; Li, L. Metabolomics and Cytokine Analysis for Identification of Severe Drug-Induced Liver Injury. J. Proteome Res. 2019, 18, 2514–2524. [Google Scholar] [CrossRef]
- Saito, K.; Kagawa, T.; Tsuji, K.; Kumagai, Y.; Sato, K.; Sakisaka, S.; Sakamoto, N.; Aiso, M.; Hirose, S.; Mori, N.; et al. Plasma Lipid Profiling of Three Types of Drug-Induced Liver Injury in Japanese Patients: A Preliminary Study. Metabolites 2020, 10, 355. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, X.; Zhang, Z.-T.; Chen, S.-S.; Li, S.-S.; Shi, Z.; Jing, J.; Huang, A.; Guo, Y.-M.; Bai, Z.-F.; et al. Metabolomics Profiling and Diagnosis Biomarkers Searching for Drug-Induced Liver Injury Implicated to Polygonum multiflorum: A Cross-Sectional Cohort Study. Front. Med. 2020, 7, 858. [Google Scholar] [CrossRef]
- Chen, S.-S.; Huang, Y.; Guo, Y.-M.; Li, S.-S.; Shi, Z.; Niu, M.; Zou, Z.-S.; Xiao, X.-H.; Wang, J.-B. Serum Metabolomic Analysis of Chronic Drug-Induced Liver Injury with or without Cirrhosis. Front. Med. 2021, 8, 640799. [Google Scholar] [CrossRef]
- Quintás, G.; Martínez-Sena, T.; Conde, I.; Ibars, E.P.; Kleinjans, J.; Castell, J.V. Metabolomic analysis to discriminate drug-induced liver injury (DILI) phenotypes. Arch. Toxicol. 2021, 95, 3049–3062. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, L.; Chen, E.; Lu, J.; Xiao, L.; Liu, Q.; Zhu, D.; Zhang, F.; Xu, X.; Li, L. Targeted Metabolomics Analysis of Bile Acids in Patients with Idiosyncratic Drug-Induced Liver Injury. Metabolites 2021, 11, 852. [Google Scholar] [CrossRef]
- Soga, T.; Sugimoto, M.; Honma, M.; Mori, M.; Igarashi, K.; Kashikura, K.; Ikeda, S.; Hirayama, A.; Yamamoto, T.; Yoshida, H.; et al. Serum metabolomics reveals γ-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J. Hepatol. 2011, 55, 896–905. [Google Scholar] [CrossRef]
- Kim, J.W.; Ryu, S.H.; Kim, S.; Lee, H.W.; Lim, M.-S.; Seong, S.J.; Kim, S.; Yoon, Y.-R.; Kim, K.-B. Pattern Recognition Analysis for Hepatotoxicity Induced by Acetaminophen Using Plasma and Urinary 1H NMR-Based Metabolomics in Humans. Anal. Chem. 2013, 85, 11326–11334. [Google Scholar] [CrossRef] [PubMed]
- Prescott, L.F.; Wright, N. The effects of hepatic and renal damage on paracetamol metabolism and excretion following overdosage.: A pharmacokinetic study. J. Cereb. Blood Flow Metab. 1973, 49, 602–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuhara, K.; Ohno, A.; Ando, Y.; Yamoto, T.; Okuda, H. A 1H NMR-based Metabolomics Approach for Mechanistic Insight into Acetaminophen-induced Hepatotoxicity. Drug Metab. Pharmacokinet. 2011, 26, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coen, M.; Lenz, E.M.; Nicholson, J.K.; Wilson, I.D.; Pognan, A.F.; Lindon, J.C. An Integrated Metabonomic Investigation of Acetaminophen Toxicity in the Mouse Using NMR Spectroscopy. Chem. Res. Toxicol. 2003, 16, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Dargue, R.; Zia, R.; Lau, C.; Nicholls, A.W.; Dare, T.O.; Lee, K.; Jalan, R.; Coen, M.; Wilson, I.D. Metabolism and Effects on Endogenous Metabolism of Paracetamol (Acetaminophen) in a Porcine Model of Liver Failure. Toxicol. Sci. 2020, 175, 87–97. [Google Scholar] [CrossRef]
- Bernal, W.; Donaldson, N.; Wyncoll, D.; Wendon, J. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: A cohort study. Lancet 2002, 359, 558–563. [Google Scholar] [CrossRef]
- Hemming, A.W.; Gallinger, S.; Greig, P.D.; Cattral, M.S.; Langer, B.; Taylor, B.R.; Verjee, Z.; Giesbrecht, E.; Nakamachi, Y.; Furuya, K.N. The hippurate ratio as an indicator of functional hepatic reserve for resection of hepatocellular carcinoma in cirrhotic patients. J. Gastrointest. Surg. 2001, 5, 316–321. [Google Scholar] [CrossRef]
- Krähenbühl, L.; Ledermann, M.; Lang, C.; Krähenbühl, S. Relationship between hepatic mitochondrial functions in vivo and in vitro in rats with carbon tetrachloride-induced liver cirrhosis. J. Hepatol. 2000, 33, 216–223. [Google Scholar] [CrossRef]
- Sun, J.; Schnackenberg, L.K.; Holland, R.D.; Schmitt, T.C.; Cantor, G.H.; Dragan, Y.P.; Beger, R.D. Metabonomics evaluation of urine from rats given acute and chronic doses of acetaminophen using NMR and UPLC/MS. J. Chromatogr. B 2008, 871, 328–340. [Google Scholar] [CrossRef]
- Ji, H.-F.; Shen, L. Uric acid potentially links fatty liver and high blood pressure. Hepatology 2010, 52, 1518–1519. [Google Scholar] [CrossRef]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azer, S.A.; Hasanato, R. Use of bile acids as potential markers of liver dysfunction in humans: A systematic review. Medicine 2021, 100, e27464. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Shafi, A.; Nguyen, T.; Draghici, S. Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biol. 2019, 20, 203. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Torres, M.; Kumar, M.; García-Llorens, G.; Quintás, G.; Tricot, T.; Boon, R.; Tolosa, L.; Toprakhisar, B.; Chesnais, F.; Verfaillie, C.; et al. A Novel UPLC-MS Metabolomic Analysis-Based Strategy to Monitor the Course and Extent of iPSC Differentiation to Hepatocytes. J. Proteome Res. 2022, 21, 702–712. [Google Scholar] [CrossRef]
- García-Cañaveras, J.C.; Jiménez, N.; Gómez-Lechón, M.J.; Castell, J.V.; Donato, M.T.; Lahoz, A. LC-MS untargeted metabolomic analysis of drug-induced hepatotoxicity in HepG2 cells. Electrophoresis 2015, 36, 2294–2302. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Torres, M.; Quintás, G.; Castell, J.V. The Potential Role of Metabolomics in Drug-Induced Liver Injury (DILI) Assessment. Metabolites 2022, 12, 564. https://doi.org/10.3390/metabo12060564
Moreno-Torres M, Quintás G, Castell JV. The Potential Role of Metabolomics in Drug-Induced Liver Injury (DILI) Assessment. Metabolites. 2022; 12(6):564. https://doi.org/10.3390/metabo12060564
Chicago/Turabian StyleMoreno-Torres, Marta, Guillermo Quintás, and José V. Castell. 2022. "The Potential Role of Metabolomics in Drug-Induced Liver Injury (DILI) Assessment" Metabolites 12, no. 6: 564. https://doi.org/10.3390/metabo12060564