
LDL Promotes Disorders in β-Cell Cholesterol Metabolism, Implications on Insulin Cellular Communication Mediated by EVs

 ,
,  ,
,  ,
,
Abstract
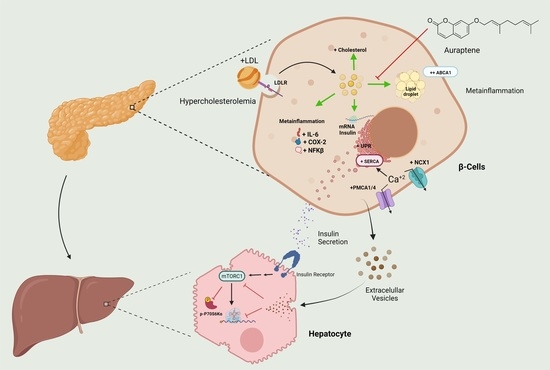
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Cultures
2.3. LDL Isolation and Fluorescent Labeling
2.4. Cell Cytometer Assays
2.5. Confocal Microscopy
2.6. Western Blot (WB) Analysis
2.7. Cellular Fractionation
2.8. Isolation of Lipid Droplets and Lipid Quantification
2.9. Insulin ELISA Assays
2.10. Quantitative PCR for Insulin
2.11. Intracellular Calcium Quantification
2.12. Auraptene Treatment
2.13. Isolation of EVs from Cell Culture-Conditioned Medium
2.14. Extracellular Vesicle Analysis
2.15. MTT Assay
2.16. Organelle Fractionation
2.17. Molecular Docking Experimentation
2.18. Statistical Analysis
3. Results
3.1. Characterization of LDL Internalization in Insulin-Secreting Cells and Expression of Cholesterol Protein Targets
3.2. Insulin Secretion Is Promoted by LDL Endocytosis and Regulation of Targets That Modulate Intracellular Calcium
3.3. Evaluation of UPR Activation and Inflammatory Pathway
3.4. Effect of LDL Treatment on Renin–Angiotensin System (RAS) Components
3.5. Intracellular Cholesterol Accumulation Induces Transporter Expression
3.6. Regulation of Intracellular Cholesterol Metabolism by Auraptene
3.7. Cellular Communication Mediated by EVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reddy, P.; Lent-Schochet, D.; Ramakrishnan, N.; McLaughlin, M.; Jialal, I. Metabolic syndrome is an inflammatory disorder: A conspiracy between adipose tissue and phagocytes. Clin. Chim. Acta 2019, 496, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Kruit, J.K.; Verchere, C.B.; Hayden, M.R. Cholesterol in islet dysfunction and type 2 diabetes. J. Clin. Investig. 2008, 118, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Hirose, H.; Ohneda, M.; Johnson, J.H.; McGarry, J.D.; Unger, R.H. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: Impairment in adipocyte-beta-cell relationships. Proc. Natl. Acad. Sci. USA 1994, 91, 10878–10882. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Hanson, R.L.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: Evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 2000, 49, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Parekh, V.S.; Reilly, S.M.; Nayak, J.; Chen, Z.; Lebeaupin, C.; Jang, I.; Zhang, J.; Prakash, T.P.; Sun, H.; et al. Chop/Ddit3 depletion in beta cells alleviates ER stress and corrects hepatic steatosis in mice. Sci. Transl. Med. 2021, 13, eaba9796. [Google Scholar] [CrossRef]
- Kouvari, M.; Chrysohoou, C.; Skoumas, J.; Pitsavos, C.; Panagiotakos, D.B.; Mantzoros, C.S.; ATTICA Study Investigators. The presence of NAFLD influences the transition of metabolically healthy to metabolically unhealthy obesity and the ten-year cardiovascular disease risk: A population-based cohort study. Metabolism 2022, 128, 154893. [Google Scholar] [CrossRef]
- Aso, Y.; Wakabayashi, S.; Yamamoto, R.; Matsutomo, R.; Takebayashi, K.; Inukai, T. Metabolic syndrome accompanied by hypercholesterolemia is strongly associated with proinflammatory state and impairment of fibrinolysis in patients with type 2 diabetes: Synergistic effects of plasminogen activator inhibitor-1 and thrombin-activatable fibrinolysis inhibitor. Diabetes Care 2005, 28, 2211–2216. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef]
- Corbett, E.F.; Michalak, K.M.; Oikawa, K.; Johnson, S.; Campbell, I.D.; Eggleton, P.; Kay, C.; Michalak, M. The conformation of calreticulin is influenced by the endoplasmic reticulum luminal environment. J. Biol. Chem. 2000, 275, 27177–27185. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2022. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Villanueva, J.F.; Diaz-Molina, R.; Garcia-Gonzalez, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Axmann, M.; Strobl, W.M.; Plochberger, B.; Stangl, H. Cholesterol transfer at the plasma membrane. Atherosclerosis 2019, 290, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Kruit, J.K.; Pape, T.D.; Timmins, J.M.; Reuwer, A.Q.; Vasanji, Z.; Marsh, B.J.; Rodrigues, B.; Johnson, J.D.; Parks, J.S.; et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat. Med. 2007, 13, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Fryirs, M.; Barter, P.J.; Rye, K.A. Cholesterol metabolism and pancreatic beta-cell function. Curr. Opin. Lipidol. 2009, 20, 159–164. [Google Scholar] [CrossRef]
- Acosta-Montano, P.; Rodriguez-Velazquez, E.; Ibarra-Lopez, E.; Frayde-Gomez, H.; Mas-Oliva, J.; Delgado-Coello, B.; Rivero, I.A.; Alatorre-Meda, M.; Aguilera, J.; Guevara-Olaya, L.; et al. Fatty Acid and Lipopolysaccharide Effect on Beta Cells Proteostasis and its Impact on Insulin Secretion. Cells 2019, 8, 884. [Google Scholar] [CrossRef]
- Wang, W.A.; Agellon, L.B.; Michalak, M. Endoplasmic reticulum calcium dictates the distribution of intracellular unesterified cholesterol. Cell Calcium 2018, 76, 116–121. [Google Scholar] [CrossRef]
- Marmugi, A.; Parnis, J.; Chen, X.; Carmichael, L.; Hardy, J.; Mannan, N.; Marchetti, P.; Piemonti, L.; Bosco, D.; Johnson, P.; et al. Sorcin Links Pancreatic beta-Cell Lipotoxicity to ER Ca2+ Stores. Diabetes 2016, 65, 1009–1021. [Google Scholar] [CrossRef]
- Chhabra, K.H.; Chodavarapu, H.; Lazartigues, E. Angiotensin converting enzyme 2: A new important player in the regulation of glycemia. IUBMB Life 2013, 65, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.B.; Chhabra, K.H.; Nguyen, V.K.; Xia, H.; Lazartigues, E. The transcription factor HNF1alpha induces expression of angiotensin-converting enzyme 2 (ACE2) in pancreatic islets from evolutionarily conserved promoter motifs. Biochim. Biophys. Acta 2013, 1829, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Hernandez, O.; Villegas-Comonfort, S.; Candanedo, F.; Gonzalez-Vazquez, M.C.; Chavez-Ocana, S.; Jimenez-Villanueva, X.; Sierra-Martinez, M.; Salazar, E.P. Elevated concentration of microvesicles isolated from peripheral blood in breast cancer patients. Arch. Med. Res. 2013, 44, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xu, A. Adipose Extracellular Vesicles in Intercellular and Inter-Organ Crosstalk in Metabolic Health and Diseases. Front. Immunol. 2021, 12, 608680. [Google Scholar] [CrossRef]
- Martinez, M.C.; Andriantsitohaina, R. Extracellular Vesicles in Metabolic Syndrome. Circ. Res. 2017, 120, 1674–1686. [Google Scholar] [CrossRef]
- Fu, Q.; Li, Y.; Jiang, H.; Shen, Z.; Gao, R.; He, Y.; Liu, Y.; Xu, K.; Yang, T. Hepatocytes derived extracellular vesicles from high-fat diet induced obese mice modulate genes expression and proliferation of islet beta cells. Biochem. Biophys. Res. Commun. 2019, 516, 1159–1166. [Google Scholar] [CrossRef]
- Fernandez-Millan, E.; Guillen, C. Multi-Organ Crosstalk with Endocrine Pancreas: A Focus on How Gut Microbiota Shapes Pancreatic Beta-Cells. Biomolecules 2022, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- De Medina, P.; Genovese, S.; Paillasse, M.R.; Mazaheri, M.; Caze-Subra, S.; Bystricky, K.; Curini, M.; Silvente-Poirot, S.; Epifano, F.; Poirot, M. Auraptene is an inhibitor of cholesterol esterification and a modulator of estrogen receptors. Mol. Pharmacol. 2010, 78, 827–836. [Google Scholar] [CrossRef]
- Suski, J.M.; Lebiedzinska, M.; Wojtala, A.; Duszynski, J.; Giorgi, C.; Pinton, P.; Wieckowski, M.R. Isolation of plasma membrane-associated membranes from rat liver. Nat. Protoc. 2014, 9, 312–322. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, V.; Mas-Oliva, J. A Novel beta-adaptin/c-Myc Complex Formation Modulated by Oxidative Stress in the Control of the Cell Cycle in Macrophages and its Implication in Atherogenesis. Sci. Rep. 2017, 7, 13442. [Google Scholar] [CrossRef]
- Harris, L.L.S.; Shew, T.M.; Skinner, J.R.; Wolins, N.E. A single centrifugation method for isolating fat droplets from cells and tissues. J. Lipid. Res. 2012, 53, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Porter, R.H.; Palmer, A.M.; Croucher, M.J. Comparison of human recombinant adenosine A2B receptor function assessed by Fluo-3-AM fluorometry and microphysiometry. Br. J. Pharmacol. 2003, 138, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.A.; Bassani, R.A.; Bassani, J.W. Measuring [Ca2+] with fluorescent indicators: Theoretical approach to the ratio method. Cell Calcium 1998, 24, 17–26. [Google Scholar] [CrossRef]
- Scullion, S.M.; Gurgul-Convey, E.; Elsner, M.; Lenzen, S.; Flatt, P.R.; McClenaghan, N.H. Enhancement of homocysteine toxicity to insulin-secreting BRIN-BD11 cells in combination with alloxan. J. Endocrinol. 2012, 214, 233–238. [Google Scholar] [CrossRef]
- PubChem Compound Summary for CID 1550607, Auraptene. 2022. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Auraptene (accessed on 1 July 2022).
- Damian-Zamacona, S.; Garcia-Gonzalez, V.; Avila-Barrientos, L.P.; Delgado-Coello, B.; Reyes-Grajeda, J.P.; Mas-Oliva, J. Cell survival regulation during receptor-mediated endocytosis of chemically-modified lipoproteins associated to the formation of an Amphiphysin 2 (Bin1)/c-Myc complex. Biochem. Biophys. Res. Commun. 2018, 505, 365–371. [Google Scholar] [CrossRef]
- Weiss, M.; Steiner, D.F.; Philipson, L.H. Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships. MDText.com, Inc., South Dartmouth (MA). 2000. Available online: https://europepmc.org/article/nbk/nbk279029 (accessed on 1 July 2022).
- Guo, W.; Gong, Y.; Fu, Z.; Fu, J.; Sun, Y.; Ju, X.; Chang, Y.; Wang, W.; Zhu, X.; Gao, B.; et al. The effect of cholesteryl ester transfer protein on pancreatic beta cell dysfunction in mice. Nutr. Metab. 2016, 13, 21. [Google Scholar] [CrossRef]
- Galindo-Hernandez, O.; Cordova-Guerrero, I.; Diaz-Rubio, L.J.; Pulido-Capiz, A.; Diaz-Villanueva, J.F.; Castaneda-Sanchez, C.Y.; Serafin-Higuera, N.; Garcia-Gonzalez, V. Protein translation associated to PERK arm is a new target for regulation of metainflammation: A connection with hepatocyte cholesterol. J. Cell Biochem. 2019, 120, 4158–4171. [Google Scholar] [CrossRef]
- Jung, B.C.; Kang, S. Epigenetic regulation of inflammatory factors in adipose tissue. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2021, 1866, 159019. [Google Scholar] [CrossRef]
- Slepchenko, K.G.; Chen, S.; Counts, G.P.; Corbin, K.L.; Colvin, R.A.; Nunemaker, C.S. Synchrotron fluorescence imaging of individual mouse beta-cells reveals changes in zinc, calcium, and iron in a model of low-grade inflammation. Metallomics 2021, 13, mfab051. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Michiko, S. ER Stress, Secretory Granule Biogenesis, and Insulin. In Ultimate Guide to Insulin; Yoko Shiba, E.D.G.Z., Ed.; IntechOpen: Rijeka, Croatia, 2018. [Google Scholar] [CrossRef]
- Martinez-Navarro, I.; Diaz-Molina, R.; Pulido-Capiz, A.; Mas-Oliva, J.; Luna-Reyes, I.; Rodriguez-Velazquez, E.; Rivero, I.A.; Ramos-Ibarra, M.A.; Alatorre-Meda, M.; Garcia-Gonzalez, V. Lipid Modulation in the Formation of beta-Sheet Structures. Implications for De Novo Design of Human Islet Amyloid Polypeptide and the Impact on beta-Cell Homeostasis. Biomolecules 2020, 10, 1201. [Google Scholar] [CrossRef]
- Ali Khan, H.; Mutus, B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2014, 2, 70. [Google Scholar] [CrossRef]
- Bindom, S.M.; Lazartigues, E. The sweeter side of ACE2: Physiological evidence for a role in diabetes. Mol. Cell Endocrinol. 2009, 302, 193–202. [Google Scholar] [CrossRef]
- Niu, M.J.; Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Loss of angiotensin-converting enzyme 2 leads to impaired glucose homeostasis in mice. Endocrine 2008, 34, 56–61. [Google Scholar] [CrossRef]
- Dong, B.; Li, H.; Singh, A.B.; Cao, A.; Liu, J. Inhibition of PCSK9 transcription by berberine involves down-regulation of hepatic HNF1alpha protein expression through the ubiquitin-proteasome degradation pathway. J. Biol. Chem. 2015, 290, 4047–4058. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109.e1015. [Google Scholar] [CrossRef]
- Muller, J.A.; Gross, R.; Conzelmann, C.; Kruger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat. Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef]
- Gkogkou, E.; Barnasas, G.; Vougas, K.; Trougakos, I.P. Expression profiling meta-analysis of ACE2 and TMPRSS2, the putative anti-inflammatory receptor and priming protease of SARS-CoV-2 in human cells, and identification of putative modulators. Redox Biol. 2020, 36, 101615. [Google Scholar] [CrossRef] [PubMed]
- Lyu, K.; Zhang, D.; Song, J.; Li, X.; Perry, R.J.; Samuel, V.T.; Shulman, G.I. Short-term overnutrition induces white adipose tissue insulin resistance through sn-1,2-diacylglycerol/PKCepsilon/insulin receptor Thr1160 phosphorylation. JCI Insight 2021, 6, e139946. [Google Scholar] [CrossRef]
- Rogers, M.A.; Liu, J.; Song, B.L.; Li, B.L.; Chang, C.C.; Chang, T.Y. Acyl-CoA:cholesterol acyltransferases (ACATs/SOATs): Enzymes with multiple sterols as substrates and as activators. J. Steroid Biochem. Mol. Biol. 2015, 151, 102–107. [Google Scholar] [CrossRef]
- Miyazaki, A.; Sakai, M.; Sakamoto, Y.; Horiuchi, S. Acyl-coenzyme A:cholesterol acyltransferase inhibitors for controlling hypercholesterolemia and atherosclerosis. Curr. Opin. Investig. Drugs 2003, 4, 1095–1099. [Google Scholar] [PubMed]
- Hafiane, A.; Gianopoulos, I.; Sorci-Thomas, M.G.; Daskalopoulou, S.S. Current models of apolipoprotein A-I lipidation by adenosine triphosphate binding cassette transporter A1. Curr. Opin. Lipidol. 2022, 33, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Akashi, S.; Morita, A.; Mochizuki, Y.; Shibuya, F.; Kamei, Y.; Miura, S. Citrus hassaku Extract Powder Increases Mitochondrial Content and Oxidative Muscle Fibers by Upregulation of PGC-1alpha in Skeletal Muscle. Nutrients 2021, 13, 497. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Meng, Q.; Fu, Y.; Yu, X.; Ji, T.; Chao, Y.; Chen, Q.; Li, Y.; Bian, H. Novel insights: Dynamic foam cells derived from the macrophage in atherosclerosis. J. Cell Physiol. 2021, 236, 6154–6167. [Google Scholar] [CrossRef]
- Korber, M.; Klein, I.; Daum, G. Steryl ester synthesis, storage and hydrolysis: A contribution to sterol homeostasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1534–1545. [Google Scholar] [CrossRef]
- Pal, P.; Gandhi, H.; Giridhar, R.; Yadav, M.R. ACAT inhibitors: The search for novel cholesterol lowering agents. Mini Rev. Med. Chem. 2013, 13, 1195–1219. [Google Scholar] [CrossRef]
- Shibuya, K.; Kawamine, K.; Miura, T.; Ozaki, C.; Edano, T.; Mizuno, K.; Yoshinaka, Y.; Tsunenari, Y. Design, synthesis and pharmacology of aortic-selective acyl-CoA: Cholesterol O-acyltransferase (ACAT/SOAT) inhibitors. Bioorg. Med. Chem. 2018, 26, 4001–4013. [Google Scholar] [CrossRef]
- Kuroyanagi, K.; Kang, M.S.; Goto, T.; Hirai, S.; Ohyama, K.; Kusudo, T.; Yu, R.; Yano, M.; Sasaki, T.; Takahashi, N.; et al. Citrus auraptene acts as an agonist for PPARs and enhances adiponectin production and MCP-1 reduction in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2008, 366, 219–225. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Jackson, C.E.; Scruggs, B.S.; Schaffer, J.E.; Hanson, P.I. Effects of Inhibiting VPS4 Support a General Role for ESCRTs in Extracellular Vesicle Biogenesis. Biophys. J. 2017, 113, 1342–1352. [Google Scholar] [CrossRef]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Ayala, A.C.-V.B.; García-González, V. Initiation of protein translation, metabolic mechanisms for cancer development, progression and chemoresistance. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2022; Volume 132. [Google Scholar]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Miani, M.; Cardozo, A.K. Signalling danger: Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013, 56, 234–241. [Google Scholar] [CrossRef]
- Lyu, J.; Fukunaga, K.; Imachi, H.; Sato, S.; Kobayashi, T.; Saheki, T.; Ibata, T.; Yoshimura, T.; Iwama, H.; Murao, K. Oxidized LDL Downregulates ABCA1 Expression via MEK/ERK/LXR Pathway in INS-1 Cells. Nutrients 2021, 13, 3017. [Google Scholar] [CrossRef]
- Wijesekara, N.; Kaur, A.; Westwell-Roper, C.; Nackiewicz, D.; Soukhatcheva, G.; Hayden, M.R.; Verchere, C.B. ABCA1 deficiency and cellular cholesterol accumulation increases islet amyloidogenesis in mice. Diabetologia 2016, 59, 1242–1246. [Google Scholar] [CrossRef]
- Bonfleur, M.L.; Vanzela, E.C.; Ribeiro, R.A.; de Gabriel Dorighello, G.; de Franca Carvalho, C.P.; Collares-Buzato, C.B.; Carneiro, E.M.; Boschero, A.C.; de Oliveira, H.C. Primary hypercholesterolaemia impairs glucose homeostasis and insulin secretion in low-density lipoprotein receptor knockout mice independently of high-fat diet and obesity. Biochim. Biophys. Acta 2010, 1801, 183–190. [Google Scholar] [CrossRef]
- Hao, M.; Head, W.S.; Gunawardana, S.C.; Hasty, A.H.; Piston, D.W. Direct effect of cholesterol on insulin secretion: A novel mechanism for pancreatic beta-cell dysfunction. Diabetes 2007, 56, 2328–2338. [Google Scholar] [CrossRef]
- Hsu, H.; Hsu, P.; Cheng, M.H.; Ito, Y.; Kanda, E.; Schaefer, E.J.; Ai, M. Lipoprotein Subfractions and Glucose Homeostasis in Prediabetes and Diabetes in Taiwan. J. Atheroscler. Thromb. 2019, 26, 890–914. [Google Scholar] [CrossRef]
- Amemiya-Kudo, M.; Oka, J.; Ide, T.; Matsuzaka, T.; Sone, H.; Yoshikawa, T.; Yahagi, N.; Ishibashi, S.; Osuga, J.; Yamada, N.; et al. Sterol regulatory element-binding proteins activate insulin gene promoter directly and indirectly through synergy with BETA2/E47. J. Biol. Chem. 2005, 280, 34577–34589. [Google Scholar] [CrossRef]
- Amemiya-Kudo, M.; Oka, J.; Takeuchi, Y.; Okazaki, H.; Yamamoto, T.; Yahagi, N.; Matsuzaka, K.; Okazaki, S.; Osuga, J.; Yamada, N.; et al. Suppression of the pancreatic duodenal homeodomain transcription factor-1 (Pdx-1) promoter by sterol regulatory element-binding protein-1c (SREBP-1c). J. Biol. Chem. 2011, 286, 27902–27914. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, A.; Gong, Y.; Sun, W.; Yan, B.; Lei, S.; Yao, L.H. Improving effect of cordycepin on insulin synthesis and secretion in normal and oxidative-damaged INS-1 cells. Eur. J. Pharmacol. 2022, 920, 174843. [Google Scholar] [CrossRef]
- Dhanya, R.; Kartha, C.C. Quercetin improves oxidative stress-induced pancreatic beta cell alterations via mTOR-signaling. Mol. Cell Biochem. 2021, 476, 3879–3887. [Google Scholar] [CrossRef]
- Urakami, T. Maturity-onset diabetes of the young (MODY): Current perspectives on diagnosis and treatment. Diabetes Metab. Syndr. Obes. 2019, 12, 1047–1056. [Google Scholar] [CrossRef]
- Yamagata, K. Roles of HNF1alpha and HNF4alpha in pancreatic beta-cells: Lessons from a monogenic form of diabetes (MODY). Vitam. Horm. 2014, 95, 407–423. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guevara-Olaya, L.; Chimal-Vega, B.; Castañeda-Sánchez, C.Y.; López-Cossio, L.Y.; Pulido-Capiz, A.; Galindo-Hernández, O.; Díaz-Molina, R.; Ruiz Esparza-Cisneros, J.; García-González, V. LDL Promotes Disorders in β-Cell Cholesterol Metabolism, Implications on Insulin Cellular Communication Mediated by EVs. Metabolites 2022, 12, 754. https://doi.org/10.3390/metabo12080754
Guevara-Olaya L, Chimal-Vega B, Castañeda-Sánchez CY, López-Cossio LY, Pulido-Capiz A, Galindo-Hernández O, Díaz-Molina R, Ruiz Esparza-Cisneros J, García-González V. LDL Promotes Disorders in β-Cell Cholesterol Metabolism, Implications on Insulin Cellular Communication Mediated by EVs. Metabolites. 2022; 12(8):754. https://doi.org/10.3390/metabo12080754
Chicago/Turabian StyleGuevara-Olaya, Lizbeth, Brenda Chimal-Vega, César Yahel Castañeda-Sánchez, Leslie Y. López-Cossio, Angel Pulido-Capiz, Octavio Galindo-Hernández, Raúl Díaz-Molina, Josefina Ruiz Esparza-Cisneros, and Victor García-González. 2022. "LDL Promotes Disorders in β-Cell Cholesterol Metabolism, Implications on Insulin Cellular Communication Mediated by EVs" Metabolites 12, no. 8: 754. https://doi.org/10.3390/metabo12080754