
Integrative Analysis of Metabolome and Transcriptome Reveals the Role of Strigolactones in Wounding-Induced Rice Metabolic Re-Programming


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Metabolic Sample Preparation and Metabolite Profiling
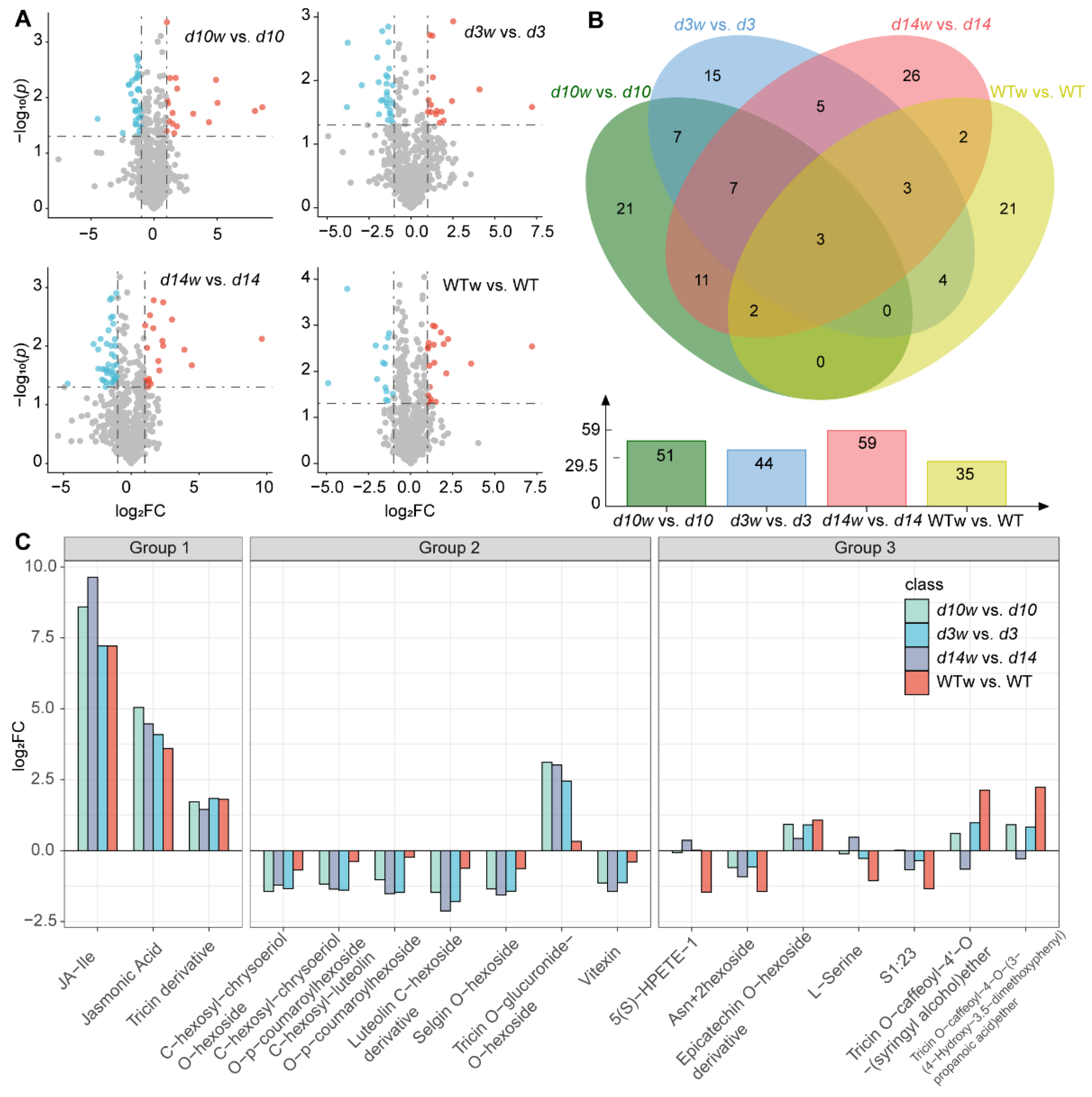
2.3. The Analysis of Differntially Accumulated Metabolites (DAMs)
2.4. RNA Sequencing
2.5. RNA-Sequencing Data Analyses
2.6. Gene Network Analysis
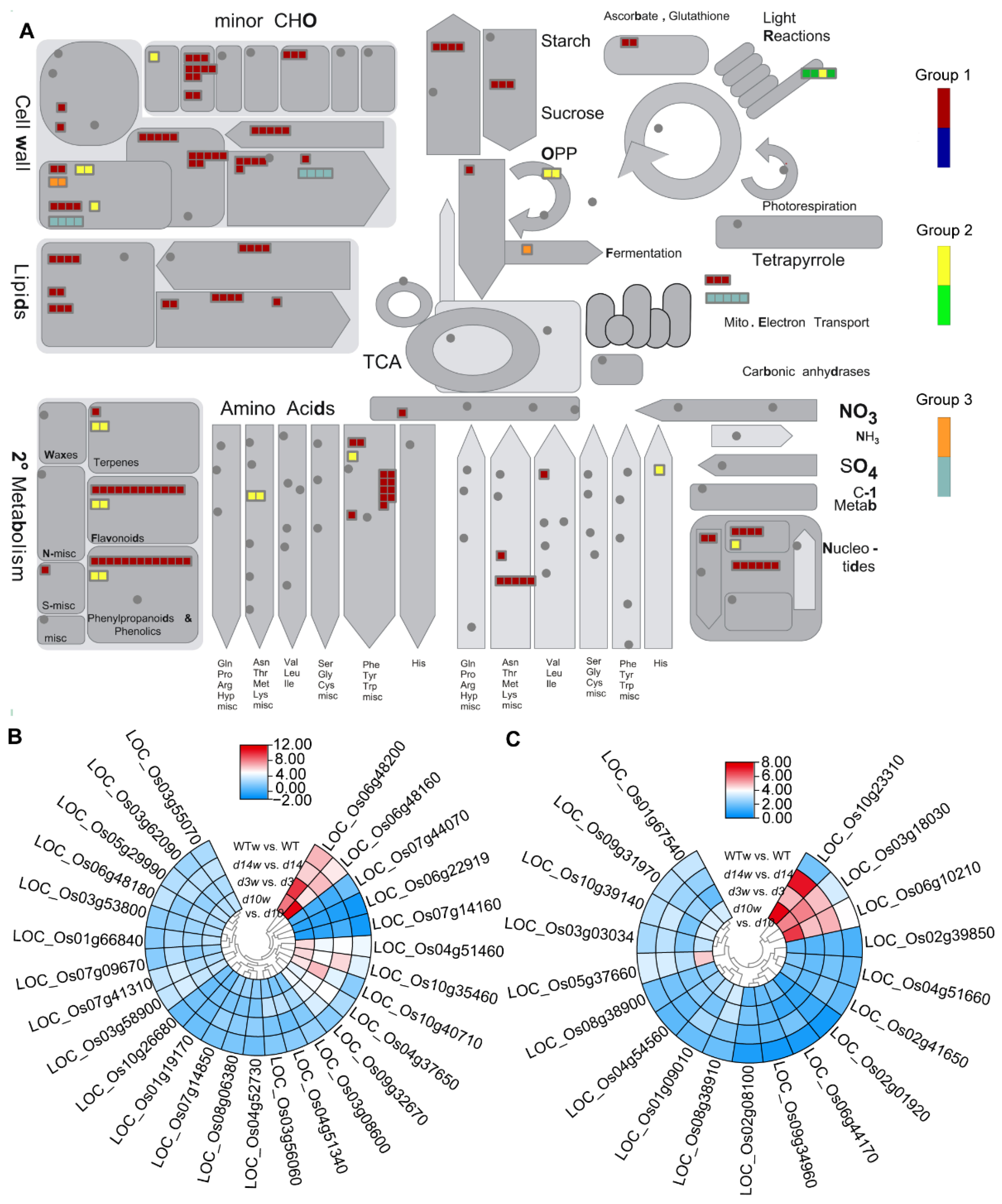
2.7. MapMan Analysis
2.8. Correlation Analysis of DEGs
3. Results
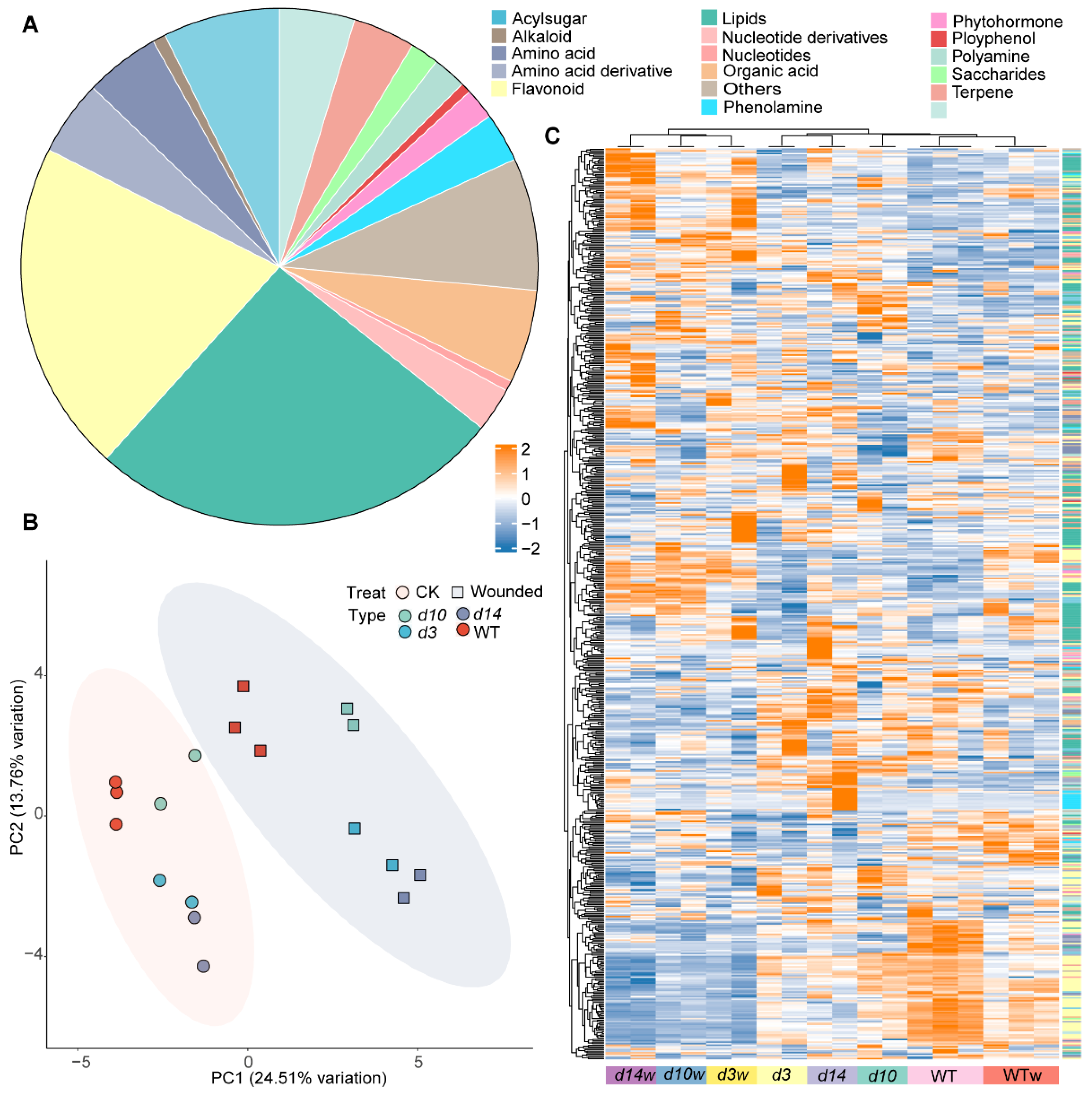
3.1. HPLC-MS/MS-Based Quantitative Metabolomic Analysis
3.2. Transcriptome Analysis
3.3. Pathway Enrichment Analysis
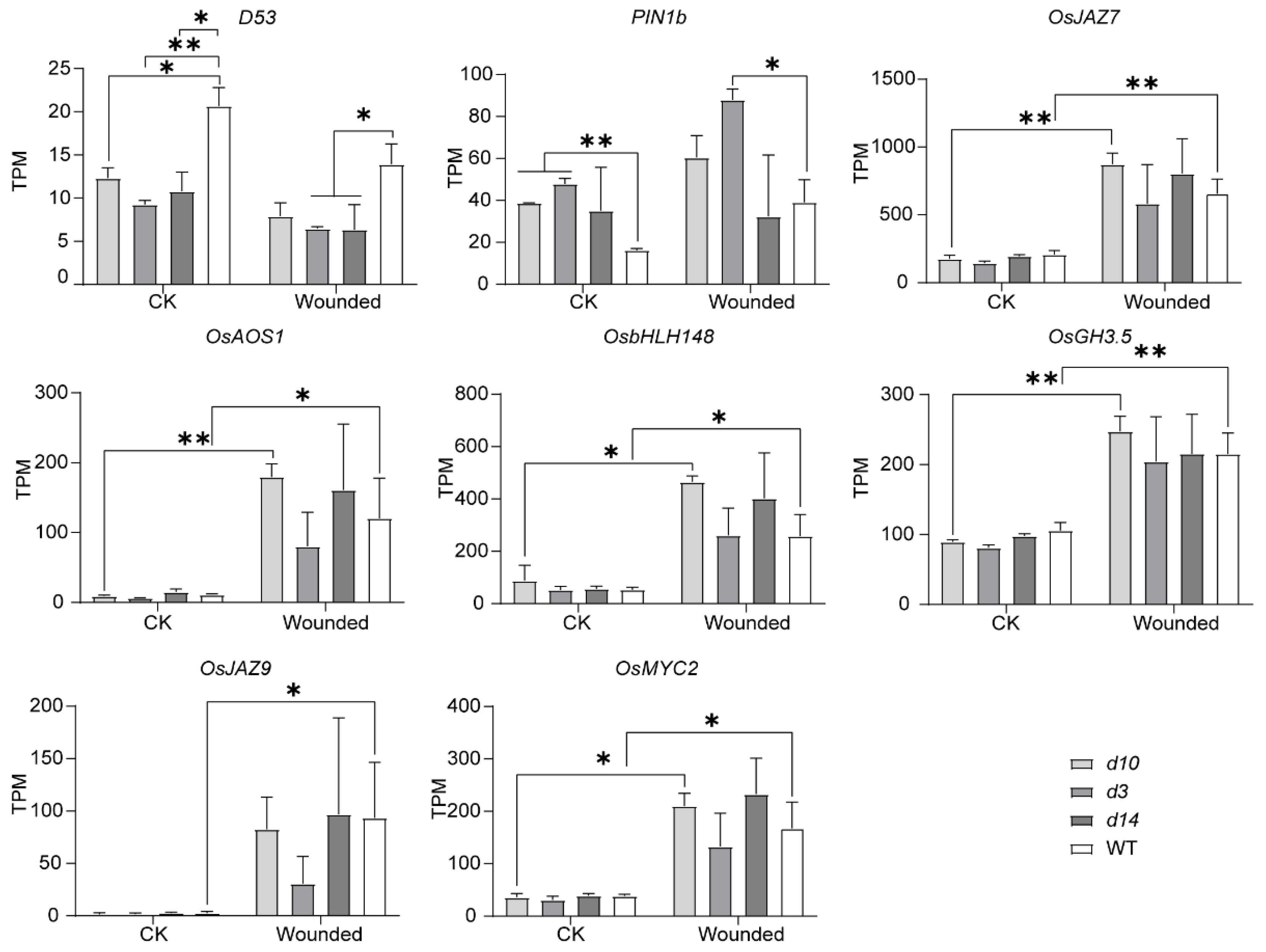
3.4. Differentially Expressed Metabolism-Related Genes
3.5. Conjoint Analysis of DEGs and DAMs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suzuki, N.; Mittler, R. Reactive oxygen species-dependent wound responses in animals and plants. Free Radic. Biol. Med. 2012, 53, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Monshausen, G.B.; Bibikova, T.N.; Weisenseel, M.H.; Gilroy, S. Ca2+ regulates reactive oxygen species production and pH during mechanosensing in Arabidopsis roots. Plant Cell 2009, 21, 2341–2356. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Baldwin, I.T.; Wu, J. Two mitogen-activated protein kinase kinases, MKK1 and MEK2, are involved in wounding- and specialist lepidopteran herbivore Manduca sexta-induced responses in Nicotiana attenuata. J. Exp. Bot. 2011, 62, 4355–4365. [Google Scholar] [CrossRef]
- Ameye, M.; Allmann, S.; Verwaeren, J.; Smagghe, G.; Haesaert, G.; Schuurink, R.C.; Audenaert, K. Green leaf volatile production by plants: A meta-Analysis. New Phytol. 2018, 220, 666–683. [Google Scholar] [CrossRef]
- Niinemets, U.; Kannaste, A.; Copolovici, L. Quantitative patterns between plant volatile emissions induced by biotic stresses and the degree of damage. Front. Plant Sci. 2013, 4, 262. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Estrada, M.; Okereke, C.N.; Jiang, Y.; Talts, E.; Kaurilind, E.; Niinemets, U. Wounding-Induced VOC Emissions in Five Tropical Agricultural Species. Molecules 2021, 26, 2602. [Google Scholar] [CrossRef]
- Mostafa, S.; Wang, Y.; Zeng, W.; Jin, B. Plant Responses to Herbivory, Wounding, and Infection. Int. J. Mol. Sci. 2022, 23, 7031. [Google Scholar] [CrossRef]
- Sardans, J.; Gargallo-Garriga, A.; Perez-Trujillo, M.; Parella, T.J.; Seco, R.; Filella, I.; Penuelas, J. Metabolic responses of Quercus ilex seedlings to wounding analysed with nuclear magnetic resonance profiling. Plant Biol. 2014, 16, 395–403. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, S.; Wang, S.; Shan, W.; Wang, X.; Lin, Y.; Su, F.; Yang, Z.; Yu, X. Defensive Responses of Tea Plants (Camellia sinensis) Against Tea Green Leafhopper Attack: A Multi-Omics Study. Front. Plant Sci. 2019, 10, 1705. [Google Scholar] [CrossRef]
- Uawisetwathana, U.; Jamboonsri, W.; Bamrungthai, J.; Jitthiang, P.; Nookaew, I.; Karoonuthaisiri, N. Metabolite profiles of brown planthopper-susceptible and resistant rice (Oryza sativa) varieties associated with infestation and mechanical stimuli. Phytochemistry 2022, 194, 113044. [Google Scholar] [CrossRef]
- Koo, A.J.; Howe, G.A. The wound hormone jasmonate. Phytochemistry 2009, 70, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Howe, G.A.; Major, I.T.; Koo, A.J. Modularity in Jasmonate Signaling for Multistress Resilience. Annu. Rev. Plant Biol. 2018, 69, 387–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Guo, Q.; Froehlich, J.E.; Hersh, H.L.; Zienkiewicz, A.; Howe, G.A.; Benning, C. Two Abscisic Acid-Responsive Plastid Lipase Genes Involved in Jasmonic Acid Biosynthesis in Arabidopsis thaliana. Plant Cell 2018, 30, 1006–1022. [Google Scholar] [CrossRef] [PubMed]
- Canonne, J.; Froidure-Nicolas, S.; Rivas, S. Phospholipases in action during plant defense signaling. Plant Signal. Behav. 2011, 6, 13–18. [Google Scholar] [CrossRef]
- Sinha, K.; Kaur, R.; Singh, N.; Kaur, S.; Rishi, V.; Bhunia, R.K. Mobilization of storage lipid reserve and expression analysis of lipase and lipoxygenase genes in rice (Oryza sativa var. Pusa Basmati 1) bran during germination. Phytochemistry 2020, 180, 112538. [Google Scholar] [CrossRef]
- Suza, W.P.; Staswick, P.E. The role of JAR1 in Jasmonoyl-L: -isoleucine production during Arabidopsis wound response. Planta 2008, 227, 1221–1232. [Google Scholar] [CrossRef]
- Hui, S.; Zhang, M.; Hao, M.; Yuan, M. Rice group I GH3 gene family, positive regulators of bacterial pathogens. Plant Signal. Behav. 2019, 14, e1588659. [Google Scholar] [CrossRef]
- Kang, J.N.; Lee, W.H.; Won, S.Y.; Chang, S.; Hong, J.P.; Oh, T.J.; Lee, S.M.; Kang, S.H. Systemic Expression of Genes Involved in the Plant Defense Response Induced by Wounding in Senna tora. Int. J. Mol. Sci. 2021, 22, 10073. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, G.; Yuan, S.; Duan, W.; Wang, P.; Bai, J.; Zhang, F.; Gao, S.; Zhang, L.; Zhao, C. TaOPR2 encodes a 12-oxo-phytodienoic acid reductase involved in the biosynthesis of jasmonic acid in wheat (Triticum aestivum L.). Biochem. Biophys. Res. Commun. 2016, 470, 233–238. [Google Scholar] [CrossRef]
- De Vleesschauwer, D.; Xu, J.; Hofte, M. Making sense of hormone-mediated defense networking: From rice to Arabidopsis. Front. Plant Sci. 2014, 5, 611. [Google Scholar] [CrossRef]
- Howles, P.A.; Gebbie, L.K.; Collings, D.A.; Varsani, A.; Broad, R.C.; Ohms, S.; Birch, R.J.; Cork, A.H.; Arioli, T.; Williamson, R.E. A temperature-sensitive allele of a putative mRNA splicing helicase down-regulates many cell wall genes and causes radial swelling in Arabidopsis thaliana. Plant Mol. Biol. 2016, 91, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, C.M.; Van der Does, D.; Zamioudis, C.; Leon-Reyes, A.; Van Wees, S.C. Hormonal modulation of plant immunity. Annu. Rev. Cell Dev. Biol. 2012, 28, 489–521. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Ma, B.; Lu, X.; Huang, Y.H.; He, S.J.; Yang, C.; Yin, C.C.; Zhao, H.; Zhou, Y.; Zhang, W.K.; et al. Ethylene-Inhibited Jasmonic Acid Biosynthesis Promotes Mesocotyl/Coleoptile Elongation of Etiolated Rice Seedlings. Plant Cell 2017, 29, 1053–1072. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Zhao, Y.; Tao, R.; Yin, L.; Gao, L.; Strid, A.; Qian, M.; Li, J.; Li, Y.; Shen, J.; et al. Ethylene mediates the branching of the jasmonate-induced flavonoid biosynthesis pathway by suppressing anthocyanin biosynthesis in red Chinese pear fruits. Plant Biotechnol. J. 2020, 18, 1223–1240. [Google Scholar] [CrossRef]
- Petrulova, V.; Vilkova, M.; Kovalikova, Z.; Sajko, M.; Repcak, M. Ethylene Induction of Non-Enzymatic Metabolic Antioxidants in Matricaria chamomilla. Molecules 2020, 25, 5720. [Google Scholar] [CrossRef]
- Alder, A.; Jamil, M.; Marzorati, M.; Bruno, M.; Vermathen, M.; Bigler, P.; Ghisla, S.; Bouwmeester, H.; Beyer, P.; Al-Babili, S. The path from β-carotene to carlactone, a strigolactone-like plant hormone. Science 2012, 335, 1348–1351. [Google Scholar] [CrossRef]
- Jia, K.P.; Baz, L.; Al-Babili, S. From carotenoids to strigolactones. J. Exp. Bot. 2018, 69, 2189–2204. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, X.; Xiong, G.; Liu, H.; Chen, F.; Wang, L.; Meng, X.; Liu, G.; Yu, H.; Yuan, Y.; et al. DWARF 53 acts as a repressor of strigolactone signalling in rice. Nature 2013, 504, 401–405. [Google Scholar] [CrossRef]
- Zhou, F.; Lin, Q.; Zhu, L.; Ren, Y.; Zhou, K.; Shabek, N.; Wu, F.; Mao, H.; Dong, W.; Gan, L.; et al. D14-SCF(D3)-dependent degradation of D53 regulates strigolactone signalling. Nature 2013, 504, 406–410. [Google Scholar] [CrossRef]
- Wu, F.; Gao, Y.; Yang, W.; Sui, N.; Zhu, J. Biological Functions of Strigolactones and Their Crosstalk With Other Phytohormones. Front. Plant. Sci. 2022, 13, 821563. [Google Scholar] [CrossRef]
- Ha, C.V.; Leyva-Gonzalez, M.A.; Osakabe, Y.; Tran, U.T.; Nishiyama, R.; Watanabe, Y.; Tanaka, M.; Seki, M.; Yamaguchi, S.; Dong, N.V.; et al. Positive regulatory role of strigolactone in plant responses to drought and salt stress. Proc. Natl. Acad. Sci. USA 2014, 111, 851–856. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, L.; Li, Y.; Li, K.; Liu, X.; Zhou, J.; Yang, C.; Liu, X.; Fang, C.; Luo, J. Integrative Metabolomic and Transcriptomic Analyses Reveal Metabolic Changes and Its Molecular Basis in Rice Mutants of the Strigolactone Pathway. Metabolites 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Joo, Y.; Cao, D.; Li, R.; Lee, G.; Halitschke, R.; Baldwin, G.; Baldwin, I.T.; Wang, M. Strigolactone signaling regulates specialized metabolism in tobacco stems and interactions with stem-feeding herbivores. PLoS Biol. 2020, 18, e3000830. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, X.; Li, K.; Wang, D.; Ding, Y.; Liu, X.; Luo, J.; Fang, C. A simple and efficient cloning system for CRISPR/Cas9-mediated genome editing in rice. PeerJ 2020, 8, e8491. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Li, K.; Wu, Y.; Wang, D.; Zhou, J.; Liu, X.; Li, Y.; Jin, C.; Liu, X.; Mur, L.A.J.; et al. OsTSD2-mediated cell wall modification affects ion homeostasis and salt tolerance. Plant Cell Environ. 2019, 42, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Dresen, S.; Ferreiros, N.; Gnann, H.; Zimmermann, R.; Weinmann, W. Detection and identification of 700 drugs by multi-target screening with a 3200 Q TRAP LC-MS/MS system and library searching. Anal. Bioanal. Chem. 2010, 396, 2425–2434. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef]
- Matsuda, F.; Okazaki, Y.; Oikawa, A.; Kusano, M.; Nakabayashi, R.; Kikuchi, J.; Yonemaru, J.; Ebana, K.; Yano, M.; Saito, K. Dissection of genotype-phenotype associations in rice grains using metabolome quantitative trait loci analysis. Plant J. 2012, 70, 624–636. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, L.; Yuan, M. Update on the Roles of Rice MAPK Cascades. Int. J. Mol. Sci. 2021, 22, 1679. [Google Scholar] [CrossRef]
- Wan, S.; Xin, X.F. Regulation and integration of plant jasmonate signaling: A comparative view of monocot and dicot. J. Genet. Genom. 2022, 49, 704–714. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; Tang, L.; Tong, X.; Wang, L.; Liu, L.; Huang, S.; Zhang, J. SAPK10-Mediated Phosphorylation on WRKY72 Releases Its Suppression on Jasmonic Acid Biosynthesis and Bacterial Blight Resistance. iScience 2019, 16, 499–510. [Google Scholar] [CrossRef]
- Shen, S.; Peng, M.; Fang, H.; Wang, Z.; Zhou, S.; Jing, X.; Zhang, M.; Yang, C.; Guo, H.; Li, Y.; et al. An Oryza-specific hydroxycinnamoyl tyramine gene cluster contributes to enhanced disease resistance. Sci. Bull. 2021, 66, 2369–2380. [Google Scholar] [CrossRef]
- Lam, P.Y.; Zhu, F.Y.; Chan, W.L.; Liu, H.; Lo, C. Cytochrome P450 93G1 Is a Flavone Synthase II That Channels Flavanones to the Biosynthesis of Tricin O-Linked Conjugates in Rice. Plant Physiol. 2014, 165, 1315–1327. [Google Scholar] [CrossRef]
- Peng, M.; Shahzad, R.; Gul, A.; Subthain, H.; Shen, S.; Lei, L.; Zheng, Z.; Zhou, J.; Lu, D.; Wang, S.; et al. Differentially evolved glucosyltransferases determine natural variation of rice flavone accumulation and UV-tolerance. Nat. Commun. 2017, 8, 1975. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Shahzad, B.; Rehman, A.; Zheng, B. Response of Phenylpropanoid Pathway and the Role of Polyphenols in Plants under Abiotic Stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Fang, P.; Zhang, H.; Chi, C.; Song, L.; Xia, X.; Shi, K.; Zhou, Y.; Zhou, J.; Yu, J. Strigolactones positively regulate defense against root-knot nematodes in tomato. J. Exp. Bot. 2019, 70, 1325–1337. [Google Scholar] [CrossRef]
- Horbowicz, M.; Wiczkowski, W.; Koczkodaj, D.; Saniewski, M. Effects of methyl jasmonate on accumulation of flavonoids in seedlings of common buckwheat (Fagopyrum esculentum Moench). Acta Biol. Hung. 2011, 62, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Elarabi, N.I.; Abdelhadi, A.A.; Sief-Eldein, A.G.M.; Ismail, I.A.; Abdallah, N.A. Overexpression of chalcone isomerase A gene in Astragalus trigonus for stimulating apigenin. Sci. Rep. 2021, 11, 24176. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Yu, H.; Guo, H.; Lin, T.; Kou, L.; Wang, A.; Shao, N.; Ma, H.; Xiong, G.; et al. Transcriptional regulation of strigolactone signalling in Arabidopsis. Nature 2020, 583, 277–281. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Li, K.; Zhou, X.; Fang, C. Integrative Analysis of Metabolome and Transcriptome Reveals the Role of Strigolactones in Wounding-Induced Rice Metabolic Re-Programming. Metabolites 2022, 12, 789. https://doi.org/10.3390/metabo12090789
Liu L, Li K, Zhou X, Fang C. Integrative Analysis of Metabolome and Transcriptome Reveals the Role of Strigolactones in Wounding-Induced Rice Metabolic Re-Programming. Metabolites. 2022; 12(9):789. https://doi.org/10.3390/metabo12090789
Chicago/Turabian StyleLiu, Ling, Kang Li, Xiujuan Zhou, and Chuanying Fang. 2022. "Integrative Analysis of Metabolome and Transcriptome Reveals the Role of Strigolactones in Wounding-Induced Rice Metabolic Re-Programming" Metabolites 12, no. 9: 789. https://doi.org/10.3390/metabo12090789
APA StyleLiu, L., Li, K., Zhou, X., & Fang, C. (2022). Integrative Analysis of Metabolome and Transcriptome Reveals the Role of Strigolactones in Wounding-Induced Rice Metabolic Re-Programming. Metabolites, 12(9), 789. https://doi.org/10.3390/metabo12090789