
Metabolomic Analysis of Wheat Grains after Tilletia laevis Kühn Infection by Using Ultrahigh-Performance Liquid Chromatography–Q-Exactive Mass Spectrometry

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strain and Spore Suspension
2.2. Metabolites Extraction for Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
2.3. Ultra-High-Performance Liquid Chromatography UHPLC-QE-MS Analysis
2.4. Data Analysis
3. Results
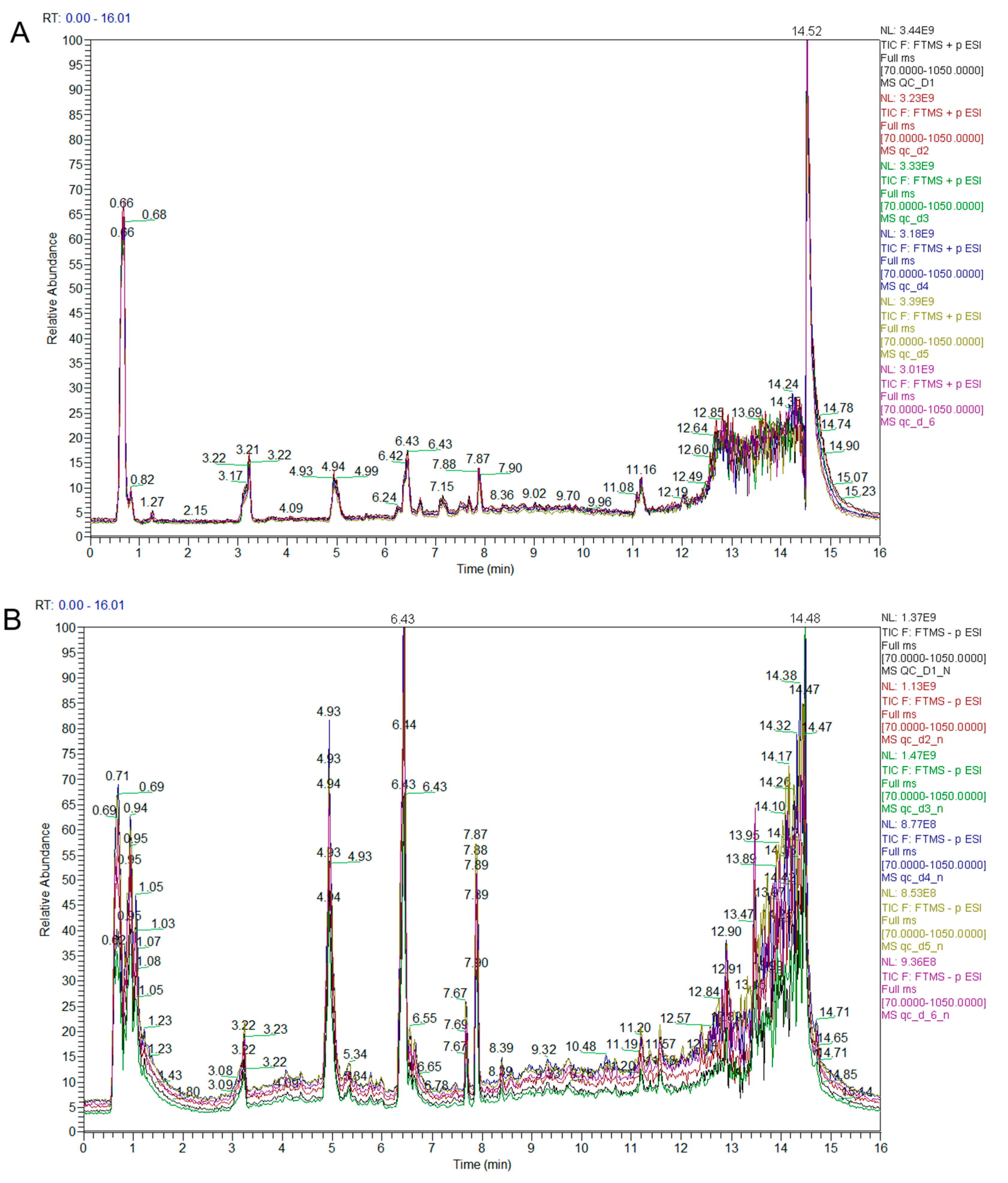
3.1. Qualitative and Quantitative Analysis of Metabolites
3.2. PCA and Orthogonal Projections to Latent Structures Discriminant Analysis
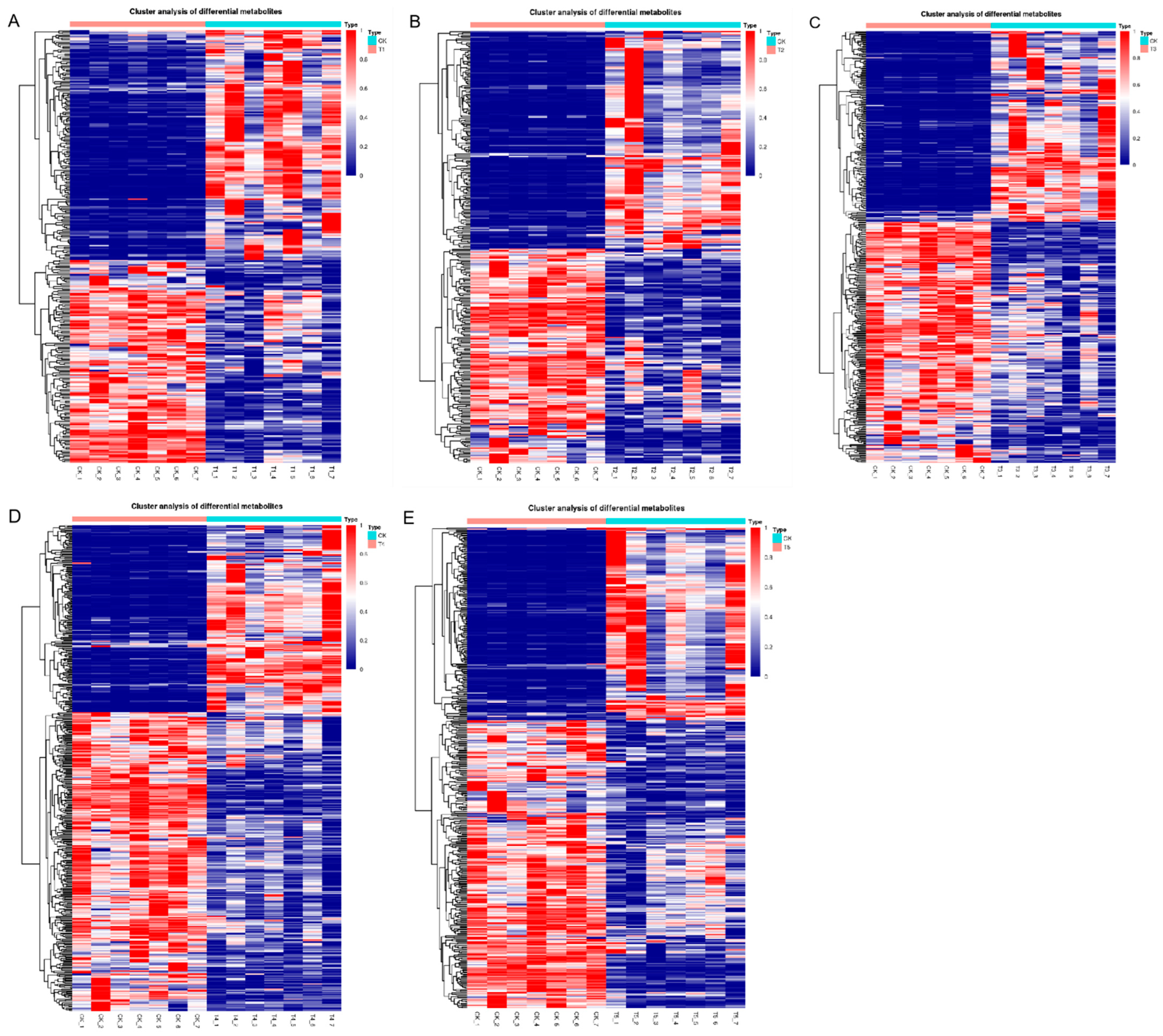
3.3. Screening and Analysis of Differential Metabolites
3.4. Analysis of Metabolic Pathways after T. laevis Infection
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonman, J.M.; Bockelman, H.E.; Goates, B.J.; Obert, D.E.; McGuire, P.E.; Qualset, C.O.; Hijmans, R.J. Geographic distribution of common and dwarf bunt resistance in landraces of Triticum aestivum subsp. aestivum. Crop Sci. 2006, 46, 1622–1629. [Google Scholar] [CrossRef]
- Din, G.M.U.; Du, Z.; Zhang, H.; Zhao, S.; Liu, T.; Chen, W.; Gao, L. Effects of Tilletia foetida on microbial communities in the rhizosphere soil of wheat seeds coated with different concentrations of Jianzhuang. Microb. Ecol. 2021, 82, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Zerucha, T.; Kim, W.K.; Mauthe, W.; Klassen, G.R. The location and nucleotide sequence of the 5S rRNA gene of bunt of wheat, Tilletia caries and T. controversa. Nucleic Acids Res. 1992, 20, 2600. [Google Scholar] [CrossRef]
- Mourad, A.M.I.; Sallam, A.; Belamkar, V.; Mahdy, E.; Bakheit, B.; Abo El-Wafaa, A.; Stephen Baenziger, P. Genetic architecture of common bunt resistance in winter wheat using genome-wide association study. BMC Plant Biol. 2018, 18, 280. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Xu, T.; Muhae-Ud-Din, G.; Guo, Q.; Liu, T.; Chen, W.; Gao, L. ITRAQ-based proteomic analysis of wheat (Triticum aestivum) spikes in response to Tilletia controversa Kühn and Tilletia foetida Kühn infection, causal organisms of dwarf bunt and common bunt of wheat. Biology 2022, 11, 865. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Ren, Z.; Muhae-Ud-Din, G.; Guo, Q.; Liu, T.; Chen, W.; Gao, L. Transcriptomics analysis of wheat tassel response to Tilletia laevis Kühn, which causes common bunt of wheat. Front. Plant Sci. 2022, 13, 823907. [Google Scholar] [CrossRef]
- Mourad, A.; Mahdy, E.; Bakheit, B.R.; Abo-Elwafaa, A.; Baenziger, P.S. Effect of common bunt infection on agronomic traits in wheat (Triticum aestivum L.). J. Plant Genet. Breed. 2018, 2, 2. [Google Scholar]
- Muhae-Ud-Din, G.; Chen, D.; Liu, T.; Chen, W.; Gao, L. Methyljasmonate and salicylic acid contribute to the control of Tilletia controversa Kühn, causal agent of wheat dwarf bunt. Sci. Rep. 2020, 10, 19175. [Google Scholar] [CrossRef]
- Gaudet, D.A.; Lu, Z.X.; Leggett, F.; Puchalski, B.; Laroche, A. Compatible and incompatible interactions in wheat involving the Bt-10 gene for resistance to Tilletia tritici, the common bunt pathogen. Phytopathology 2007, 97, 1397–1405. [Google Scholar] [CrossRef]
- Lu, Z.X.; Gaudet, D.A.; Frick, M.; Puchalski, B.; Genswein, B.; Laroche, A. Identification and characterization of genes differentially expressed in the resistance reaction in wheat infected with Tilletia tritici, the common bunt pathogen. J. Biochem. Mol. Biol. 2005, 38, 420–431. [Google Scholar] [CrossRef]
- Swinburne, T.R. Infection of wheat by Tilletia caries (DC.) TUL., the causal organism of bunt. Trans. Br. Mycol. Soc. 1963, 46, 145–156. [Google Scholar] [CrossRef]
- Fernandez, J.A.; Duran, R.; Schafer, J.F. Histological aspects of dwarf bunt resistance in wheat. Phytopathology 1978, 68, 1417–1421. [Google Scholar] [CrossRef]
- Xu, T.; Qin, D.; Muhae Ud Din, G.; Liu, T.; Chen, W.; Gao, L. Characterization of histological changes at the tillering stage (Z21) in resistant and susceptible wheat plants infected by Tilletia controversa Kühn. BMC Plant Biol. 2021, 21, 49. [Google Scholar] [CrossRef]
- Kessler, A.; Kalske, A. Plant secondary metabolite diversity and species interactions. Annu. Rev. Ecol. Evol. Syst. 2018, 49, 115–138. [Google Scholar] [CrossRef]
- Pott, D.M.; Osorio, S.; Vallarino, J.G. From central to specialized metabolism: An overview of some secondary compounds derived from the primary metabolism for their role in conferring nutritional and organoleptic characteristics to fruit. Front. Plant Sci. 2019, 10, 835. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Fernie, A.R.; Luo, J. Exploring the diversity of plant metabolism. Trends Plant Sci. 2019, 24, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Sulpice, R.; McKeown, P.C. Moving toward a comprehensive map of central plant metabolism. Annu. Rev. Plant Biol. 2015, 66, 187–210. [Google Scholar] [CrossRef]
- Castro-moretti, F.R.; Gentzel, I.N.; Mackey, D.; Alonso, A.P. Metabolomics as an emerging tool for the study of plant–pathogen interactions. Metabolites 2020, 10, 52. [Google Scholar] [CrossRef]
- Solomon, P.S.; Oliver, R.P. Evidence that γ-aminobutyric acid is a major nitrogen source during Cladosporium fulvum infection of tomato. Planta 2002, 214, 414–420. [Google Scholar] [CrossRef]
- Solomon, P.S.; Oliver, R.P. The nitrogen content of the tomato leaf apoplast increases during infection by Cladosporium fulvum. Planta 2001, 213, 241–249. [Google Scholar] [CrossRef]
- Etalo, D.W.; Stulemeijer, I.J.E.; Peter van Esse, H.; de Vos, R.C.H.; Bouwmeester, H.J.; Joosten, M.H.A.J. System-Wide hypersensitive response-associated transcriptome and metabolome reprogramming in tomato. Plant Physiol. 2013, 162, 1599–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asselin, J.E.; Lin, J.; Perez-Quintero, A.L.; Gentzel, I.; Majerczak, D.; Opiyo, S.O.; Zhao, W.; Paek, S.M.; Kim, M.G.; Coplin, D.L.; et al. Perturbation of maize phenylpropanoid metabolism by an AvrE family type III effector from Pantoea stewartii. Plant Physiol. 2015, 167, 1117–1135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lin, J.; Johnson, A.; Morgan, R.L.; Zhong, W.; Ma, W. Pseudomonas syringae type III effector HopZ1 targets a host enzyme to suppress isoflavone biosynthesis and promote infection in soybean. Cell Host Microbe 2011, 9, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Atanasova-penichon, V.; Chéreau, S.; Richard-forget, F. Metabolomics to decipher the chemical defense of cereals against Fusarium graminearum and deoxynivalenol accumulation. Int. J. Mol. Sci. 2015, 16, 24839–24872. [Google Scholar] [CrossRef] [PubMed]
- Steinmeyer, S.; Lee, K.; Jayaraman, A.; Alaniz, R.C. Microbiota metabolite regulation of host immune homeostasis: A mechanistic missing link. Curr. Allergy Asthma Rep. 2015, 15, 24. [Google Scholar] [CrossRef]
- Keon, J.; Antoniw, J.; Carzaniga, R.; Deller, S.; Ward, J.L.; Baker, J.M.; Beale, M.H.; Hammond-Kosack, K.; Rudd, J.J. Transcriptional adaptation of Mycosphaerella graminicola to programmed cell death (PCD) of its susceptible wheat host. Mol. Plant-Microbe Interact. 2007, 20, 178–193. [Google Scholar] [CrossRef]
- Copley, T.R.; Aliferis, K.A.; Kliebenstein, D.J.; Jabaji, S.H. An integrated RNAseq-1H NMR metabolomics approach to understand soybean primary metabolism regulation in response to Rhizoctonia foliar blight disease. BMC Plant Biol. 2017, 17, 84. [Google Scholar] [CrossRef]
- Song, F.; Geng, Q.; Wang, X.; Gao, X.; He, X.; Zhao, W.; Lan, H.; Tian, J.; Yang, K.; Wang, S. Gas chromatography–mass spectrometry profiling of volatile compounds reveals metabolic changes in a non-aflatoxigenic aspergillus flavus induced by 5-azacytidine. Toxins 2020, 12, 57. [Google Scholar] [CrossRef]
- Yan, S.; Liang, Y.; Zhang, J.; Chen, Z.; Liu, C.M. Autoxidated linolenic acid inhibits aflatoxin biosynthesis in Aspergillus flavus via oxylipin species. Fungal Genet. Biol. 2014, 81, 229–237. [Google Scholar] [CrossRef]
- Kshirsagar, P.R.; Hegde, H.; Pai, S.R. Storing of extracts in polypropylene microcentrifuge tubes yields contaminant peak during ultra-flow liquid chromatographic analysis. Pharmacogn. Mag. 2016, 12, S303–S306. [Google Scholar] [CrossRef]
- Muhae-Ud-Din, G.; Chen, D.; Liu, T.; Chen, W.; Gao, L. Characterization of the wheat cultivars against Tilletia controversa Kühn, causal agent of wheat dwarf bunt. Sci. Rep. 2020, 10, 9029. [Google Scholar] [CrossRef] [PubMed]
- Méchin, V.; Laluc, A.; Legée, F.; Cézard, L.; Denoue, D.; Barrière, Y.; Lapierre, C. Impact of the brown-midrib bm 5 mutation on maize lignins. J. Agric. Food Chem. 2014, 62, 5102–5107. [Google Scholar] [CrossRef] [PubMed]
- Tsogtbaatar, E.; Cocuron, J.C.; Sonera, M.C.; Alonso, A.P. Metabolite fingerprinting of pennycress (Thlaspi arvense L.) embryos to assess active pathways during oil synthesis. J. Exp. Bot. 2015, 66, 4267–4277. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant metabolomics: An indispensable system biology tool for plant science. Int. J. Mol. Sci. 2016, 17, 676. [Google Scholar] [CrossRef]
- O’Leary, B.M.; Neale, H.C.; Geilfus, C.M.; Jackson, R.W.; Arnold, D.L.; Preston, G.M. Early changes in apoplast composition associated with defence and disease in interactions between Phaseolus vulgaris and the halo blight pathogen Pseudomonas syringae Pv. phaseolicola. Plant Cell Environ. 2016, 39, 2172–2184. [Google Scholar] [CrossRef]
- Lowe-Power, T.M.; Hendrich, C.G.; von Roepenack-Lahaye, E.; Li, B.; Wu, D.; Mitra, R.; Dalsing, B.L.; Ricca, P.; Naidoo, J.; Cook, D.; et al. Metabolomics of tomato xylem sap during bacterial wilt reveals Ralstonia solanacearum produces abundant putrescine, a metabolite that accelerates wilt disease. Environ. Microbiol. 2018, 20, 1330–1349. [Google Scholar] [CrossRef]
- Duan, G.; Christian, N.; Schwachtje, J.; Walther, D.; Ebenhöh, O. The metabolic interplay between plants and phytopathogens. Metabolites 2013, 3, 1–23. [Google Scholar] [CrossRef]
- Berger, S.; Sinha, A.K.; Roitsch, T. Plant physiology meets phytopathology: Plant primary metabolism and plant-pathogen interactions. J. Exp. Bot. 2007, 58, 4019–4026. [Google Scholar] [CrossRef]
- Raghunathan, A.; Reed, J.; Shin, S.; Palsson, B.; Daefler, S. Constraint-based analysis of metabolic capacity of Salmonella typhimurium during host-pathogen interaction. BMC Syst. Biol. 2009, 3, 38. [Google Scholar] [CrossRef]
- Raghunathan, A.; Price, N.D.; Galperin, M.Y.; Makarova, K.S.; Purvine, S.; Picone, A.F.; Cherny, T.; Xie, T.; Reilly, T.J.; Munson, R.; et al. In silico metabolic model and protein expression of Haemophilus influenzae strain Rd KW20 in rich medium. OMICS J. Integr. Biol. 2004, 8, 25–41. [Google Scholar] [CrossRef]
- Igamberdiev, A.U.; Eprintsev, A.T. Organic acids: The pools of fixed carbon involved in redox regulation and energy balance in higher plants. Front. Plant Sci. 2016, 7, 1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surendra, A.; Cuperlovic-Culf, M. Database of resistance related metabolites in wheat Fusarium head blight disease (MWFD). Database 2017, 2017, bax076. [Google Scholar] [CrossRef] [PubMed]
- Morkunas, I.; Ratajczak, L. The role of sugar signaling in plant defense responses against fungal pathogens. Acta Physiol. Plant. 2014, 36, 1607–1619. [Google Scholar] [CrossRef]
- El-Azaz, J.; de la Torre, F.; Pascual, M.B.; Debille, S.; Canlet, F.; Harvengt, L.; Trontin, J.-F.; Ávila, C.; Cánovas, F.M. Transcriptional analysis of arogenate dehydratase genes identifies a link between phenylalanine biosynthesis and lignin biosynthesis. J. Exp. Bot. 2020, 71, 3080–3093. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Xiao, S.; Wang, X.; Ao, C.; Zhang, X.; Zhu, L. GhWRKY1-like enhances cotton resistance to Verticillium dahliae via an increase in defense-induced lignification and S monolignol content. Plant Sci. 2021, 305, 110833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.S.; Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 2016, 8, 46. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef]
- Burow, G.B.; Nesbitt, T.C.; Dunlap, J.; Keller, N.P. Seed lipoxygenase products modulate Aspergillus mycotoxin biosynthesis. Mol. Plant-Microbe Interact. 1997, 10, 380–387. [Google Scholar] [CrossRef]
- De Souza, J.T.; Arnould, C.; Deulvot, C.; Lemanceau, P.; Gianinazzi-Pearson, V.; Raaijmakers, J.M. Effect of 2,4-diacetylphloroglucinol on Pythium: Cellular responses and variation in sensitivity among propagules and species. Phytopathology 2003, 93, 966–975. [Google Scholar] [CrossRef]
- Boatright, J.; Negre, F.; Chen, X.; Kish, C.M.; Wood, B.; Peel, G.; Orlova, I.; Gang, D.; Rhodes, D.; Dudareva, N. Un-derstanding in vivo benzenoid metabolism in petunia petal tissue. Plant Physiol. 2011, 135, 1993–2011. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Counts | Percentage (%) |
|---|---|---|
| Organic acids and derivatives | 224 | 30.52 |
| Organoheterocyclic compounds | 170 | 23.16 |
| Lipids and lipid-like molecules | 128 | 17.44 |
| Organic nitrogen compounds | 85 | 11.58 |
| Benzenoids | 64 | 8.71 |
| Phenylpropanoids and polyketides | 32 | 4.35 |
| Nucleosides, nucleotides, and their analogues | 21 | 2.86 |
| Alkaloids and their derivatives | 10 | 1.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabran, M.; Chen, D.; Muhae-Ud-Din, G.; Liu, T.; Chen, W.; Liu, C.; Gao, L. Metabolomic Analysis of Wheat Grains after Tilletia laevis Kühn Infection by Using Ultrahigh-Performance Liquid Chromatography–Q-Exactive Mass Spectrometry. Metabolites 2022, 12, 805. https://doi.org/10.3390/metabo12090805
Jabran M, Chen D, Muhae-Ud-Din G, Liu T, Chen W, Liu C, Gao L. Metabolomic Analysis of Wheat Grains after Tilletia laevis Kühn Infection by Using Ultrahigh-Performance Liquid Chromatography–Q-Exactive Mass Spectrometry. Metabolites. 2022; 12(9):805. https://doi.org/10.3390/metabo12090805
Chicago/Turabian StyleJabran, Muhammad, Delai Chen, Ghulam Muhae-Ud-Din, Taiguo Liu, Wanquan Chen, Changzhong Liu, and Li Gao. 2022. "Metabolomic Analysis of Wheat Grains after Tilletia laevis Kühn Infection by Using Ultrahigh-Performance Liquid Chromatography–Q-Exactive Mass Spectrometry" Metabolites 12, no. 9: 805. https://doi.org/10.3390/metabo12090805
APA StyleJabran, M., Chen, D., Muhae-Ud-Din, G., Liu, T., Chen, W., Liu, C., & Gao, L. (2022). Metabolomic Analysis of Wheat Grains after Tilletia laevis Kühn Infection by Using Ultrahigh-Performance Liquid Chromatography–Q-Exactive Mass Spectrometry. Metabolites, 12(9), 805. https://doi.org/10.3390/metabo12090805