
Cardiovascular and Neuronal Consequences of Thyroid Hormones Alterations in the Ischemic Stroke
 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Deiodinases-Mediated Thyroid Hormones Metabolism in the Brain Physiology and Pathology
3. Neuroprotective Actions of Thyroid Hormones in Acute Stroke
4. Correlations between Thyroid Hormones Alterations and Stroke
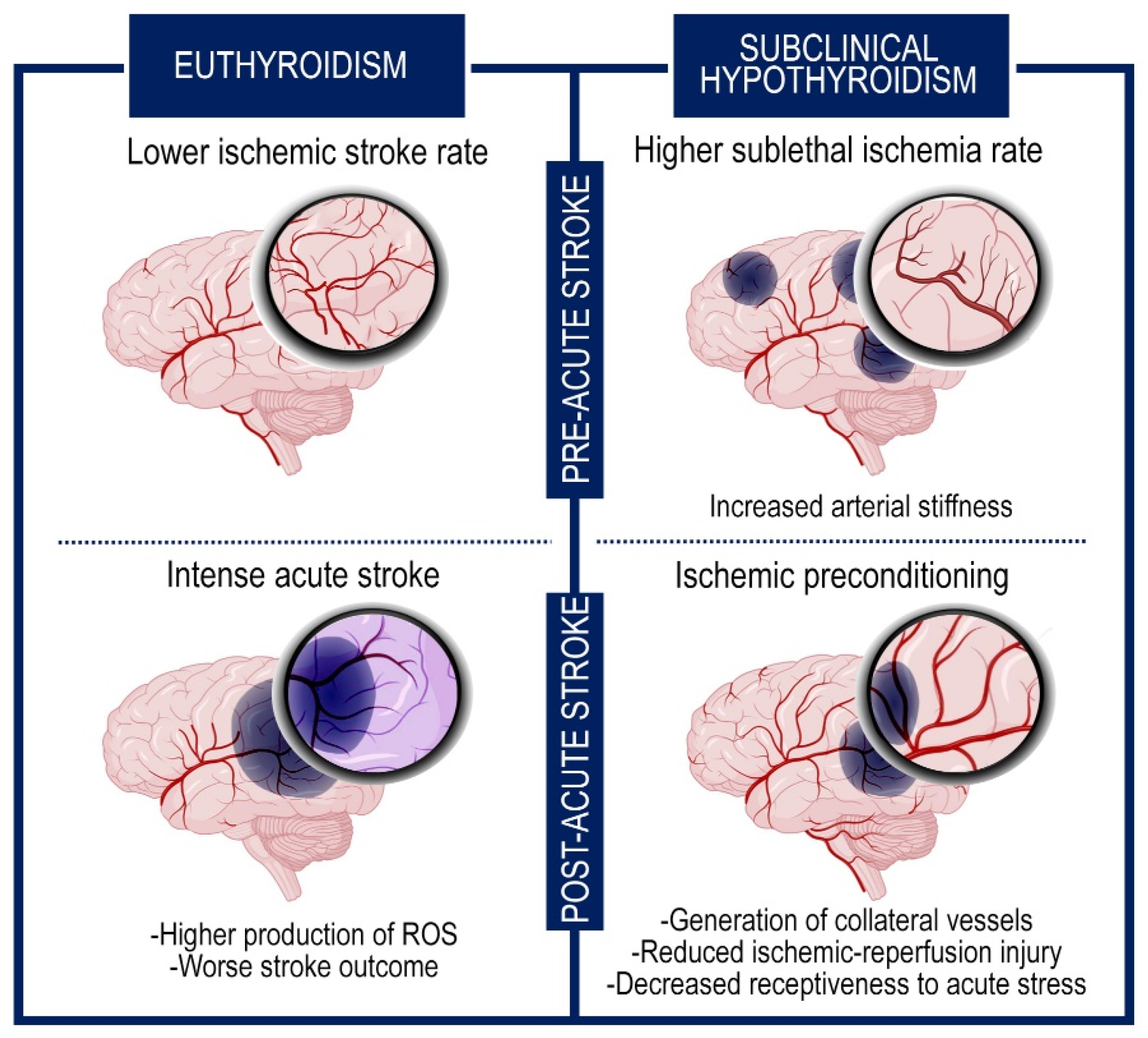
5. Cerebral Impact of Subclinical Hypothyroidism
6. Thyroid Hormones and Clinical Outcomes in Post-Stroke Patients
7. Thyroid Hormones Effects on Acute Heart Disease
8. Discussion
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shang, Y.; Zhang, Z.; Tian, J.; Li, X. Anti-Inflammatory Effects of Natural Products on Cerebral Ischemia. Front. Pharmacol. 2022, 13, 914630. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Sun, G.; Wu, Q.; Zhang, W.; Tang, M.; Zhao, T.; Hu, S. Case Report: Three-Dimensional Printing–Assisted Surgical Treatment of Complex Body Vein Ectopic Drainage. Front. Cardiovasc. Med. 2022, 9, 782601. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.L.; Adams, R.; Albers, G.; Alberts, M.J.; Benavente, O.; Furie, K.; Goldstein, L.B.; Gorelick, P.; Halperin, J.; Harbaugh, R.; et al. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: A statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: Co-sponsored by the Council on Cardiovascular Radiology and Intervention: The American Academy of Neurology affirms the value of this guideline. Circulation 2006, 113, e409–e449. [Google Scholar] [PubMed]
- Bernal-Pacheco, O.; Román, G.C. Environmental vascular risk factors: New perspectives for stroke prevention. J. Neurol. Sci. 2007, 262, 60–70. [Google Scholar] [CrossRef]
- Magid-Bernstein, J.; Girard, R.; Polster, S.; Srinath, A.; Romanos, S.; Awad, I.A.; Sansing, L.H. Cerebral Hemorrhage: Pathophysiology, Treatment, and Future Directions. Circ. Res. 2022, 130, 1204–1229. [Google Scholar] [CrossRef]
- Bahrami, M.; Keyhanifard, M.; Afzali, M. Spontaneous intracerebral hemorrhage, initial computed tomography (CT) scan findings, clinical manifestations and possible risk factors. Am. J. Nucl. Med. Mol. Imaging 2022, 12, 106–112. [Google Scholar]
- Linfante, I.; Wakhloo, A. Brain aneurysms and arteriovenous malformations: Advancements and emerging treatments in endovascular embolization. Stroke 2007, 38, 1411–1417. [Google Scholar] [CrossRef] [Green Version]
- Stapf, C.; Labovitz, D.L.; Sciacca, R.R.; Mast, H.; Mohr, J.P.; Sacco, R.L. Incidence of adult brain arteriovenous malformation hemorrhage in a prospective population-based stroke survey. Cerebrovasc. Dis. 2002, 13, 43–46. [Google Scholar] [CrossRef]
- Krishnamurthi, R.V.; Feigin, V.L.; Forouzanfar, M.H.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.M.; Truelsen, T.; et al. Global and regional burden of first-ever ischaemic and haemorrhagic stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet Glob. Health 2013, 1, e259–e281. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Jing, J.; Chen, S.; Liu, X.; Tang, Y.; Pan, C.; Tang, Z. Changes in Cerebral Blood Flow and Diffusion-Weighted Imaging Lesions after Intracerebral Hemorrhage. Transl. Stroke Res. 2022, 13, 686–706. [Google Scholar] [CrossRef]
- Robbins, N.; Swanson, R. Opposing effects of glucose on stroke and reperfusion injury: Acidosis, oxidative stress, and energy metabolism. Stroke 2014, 45, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, X.; Xu, Z.; Wang, Y.; Jiang, T.; Wang, M.; Deng, Q.; Zhou, J. Construction of a Glycaemia-Based Signature for Predicting Acute Kidney Injury in Ischaemic Stroke Patients after Endovascular Treatment. J. Clin. Med. 2022, 11, 3865. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.R.; Newhouse, J.P. The Toxic Effect of Sodium L-Glutamate on the Inner Layers of the Retina. Arch. Ophthalmol. 1957, 58, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell. Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Matuz-Mares, D.; González-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vázquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Pecoraro, R.; Arnao, V.; Maugeri, R.; Iacopino, D.G.; Pinto, A. Developing drug strategies for the neuroprotective treatment of acute ischemic stroke. Expert Rev. Neurother. 2015, 15, 1271–1284. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, S.; He, Q.; Zhang, D.; Chang, J. The Role of Immune Cells in Post-Stroke Angiogenesis and Neuronal Remodeling: The Known and the Unknown. Front. Immunol. 2021, 12, 784098. [Google Scholar] [CrossRef]
- Lambertsen, K.L.; Gregersen, R.; Meldgaard, M.; Clausen, B.H.; Heibøl, E.K.; Ladeby, R.; Knudsen, J.; Frandsen, A.; Owens, T.; Finsen, B. A role for interferon-gamma in focal cerebral ischemia in mice. J. Neuropathol. Exp. Neurol. 2004, 63, 942–955. [Google Scholar] [CrossRef] [Green Version]
- López-Valdés, H.E.; Martínez-Coria, H.; Arrieta-Cruz, I.; Cruz, M.-E. Physiopathology of ischemic stroke and its modulation using memantine: Evidence from preclinical stroke. Neural Regen. Res. 2021, 16, 433–439. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, L.M.; Conway, S.E.; Czap, A.; Malchoff, C.D.; Benashski, S.; Fortunato, G.; Staff, I.; McCullough, L.D. Thyroid hormones and functional outcomes after ischemic stroke. Thyroid Res. 2015, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Davis, P.J.; Glinsky, G.V.; Lin, H.-Y.; Mousa, S.A. Actions of Thyroid Hormone Analogues on Chemokines. J. Immunol. Res. 2016, 2016, 3147671. [Google Scholar] [CrossRef] [PubMed]
- Mancino, G.; Miro, C.; Di Cicco, E.; Dentice, M. Thyroid hormone action in epidermal development and homeostasis and its implications in the pathophysiology of the skin. J. Endocrinol. Investig. 2021, 44, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Nappi, A.; Murolo, M.; Cicatiello, A.G.; Sagliocchi, S.; Di Cicco, E.; Raia, M.; Stornaiuolo, M.; Dentice, M.; Miro, C. Thyroid Hormone Receptor Isoforms Alpha and Beta Play Convergent Roles in Muscle Physiology and Metabolic Regulation. Metabolites 2022, 12, 405. [Google Scholar] [CrossRef] [PubMed]
- Nappi, A.; Murolo, M.; Sagliocchi, S.; Miro, C.; Cicatiello, A.; Di Cicco, E.; Di Paola, R.; Raia, M.; D’Esposito, L.; Stornaiuolo, M.; et al. Selective Inhibition of Genomic and Non-Genomic Effects of Thyroid Hormone Regulates Muscle Cell Differentiation and Metabolic Behavior. Int. J. Mol. Sci. 2021, 22, 7175. [Google Scholar] [CrossRef] [PubMed]
- Visser, W.E.; Friesema, E.C.H.; Visser, T.J. Minireview: Thyroid hormone transporters: The knowns and the unknowns. Mol. Endocrinol. 2011, 25, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [Green Version]
- Gereben, B.; Zeöld, A.; Dentice, M.; Salvatore, D.; Bianco, A.C. Activation and inactivation of thyroid hormone by deiodinases: Local action with general consequences. Cell. Mol. Life Sci. 2007, 65, 570–590. [Google Scholar] [CrossRef]
- Luongo, C.; Dentice, M.; Salvatore, D. Deiodinases and their intricate role in thyroid hormone homeostasis. Nat. Rev. Endocrinol. 2019, 15, 479–488. [Google Scholar] [CrossRef]
- Miro, C.; Di Cicco, E.; Ambrosio, R.; Mancino, G.; Di Girolamo, D.; Cicatiello, A.; Sagliocchi, S.; Nappi, A.; De Stefano, M.; Luongo, C.; et al. Thyroid hormone induces progression and invasiveness of squamous cell carcinomas by promoting a ZEB-1/E-cadherin switch. Nat. Commun. 2019, 10, 5410. [Google Scholar] [CrossRef] [Green Version]
- Miro, C.; Nappi, A.; Cicatiello, A.; Di Cicco, E.; Sagliocchi, S.; Murolo, M.; Belli, V.; Troiani, T.; Albanese, S.; Amiranda, S.; et al. Thyroid Hormone Enhances Angiogenesis and the Warburg Effect in Squamous Cell Carcinomas. Cancers 2021, 13, 2743. [Google Scholar] [CrossRef]
- Dentice, M.; Marsili, A.; Zavacki, A.; Larsen, P.R.; Salvatore, D. The deiodinases and the control of intracellular thyroid hormone signaling during cellular differentiation. Biochim. Biophys. Acta 2013, 1830, 3937–3945. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Salvatore, D.; Gereben, B.; Berry, M.; Larsen, P. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [PubMed]
- Van der Geyten, S.; Segers, I.; Gereben, B.; Bartha, T.; Rudas, P.; Larsen, P.; Kühn, E.R.; Darras, V.M. Transcriptional regulation of iodothyronine deiodinases during embryonic development. Mol. Cell. Endocrinol. 2001, 183, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Di Cicco, E.; Moran, C.; Visser, W.; Nappi, A.; Schoenmakers, E.; Todd, P.; Lyons, G.; Dattani, M.; Ambrosio, R.; Parisi, S.; et al. Germ Line Mutations in the Thyroid Hormone Receptor Alpha Gene Predispose to Cutaneous Tags and Melanocytic Nevi. Thyroid 2021, 31, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Miro, C.; Di Giovanni, A.; Murolo, M.; Cicatiello, A.G.; Nappi, A.; Sagliocchi, S.; Di Cicco, E.; Morra, F.; Celetti, A.; Pacifico, F.; et al. Thyroid hormone and androgen signals mutually interplay and enhance inflammation and tumorigenic activation of tumor microenvironment in prostate cancer. Cancer Lett. 2022, 532, 215581. [Google Scholar] [CrossRef]
- Patel, J.; Landers, K.; Li, H.; Mortimer, R.H.; Richard, K. Thyroid hormones and fetal neurological development. J. Endocrinol. 2011, 209, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bath, S.C. The effect of iodine deficiency during pregnancy on child development. Proc. Nutr. Soc. 2019, 78, 150–160. [Google Scholar] [CrossRef]
- Kim, H.K.; Song, J. Hypothyroidism and Diabetes-Related Dementia: Focused on Neuronal Dysfunction, Insulin Resistance, and Dyslipidemia. Int. J. Mol. Sci. 2022, 23, 2982. [Google Scholar] [CrossRef]
- Dentice, M.; Ambrosio, R.; Damiano, V.; Sibilio, A.; Luongo, C.; Guardiola, O.; Yennek, S.; Zordan, P.; Minchiotti, G.; Colao, A.; et al. Intracellular inactivation of thyroid hormone is a survival mechanism for muscle stem cell proliferation and lineage progression. Cell Metab. 2014, 20, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Miro, C.; Ambrosio, R.; De Stefano, M.A.; Di Girolamo, D.; Di Cicco, E.; Cicatiello, A.G.; Mancino, G.; Porcelli, T.; Raia, M.; Del Vecchio, L.; et al. The Concerted Action of Type 2 and Type 3 Deiodinases Regulates the Cell Cycle and Survival of Basal Cell Carcinoma Cells. Thyroid 2017, 27, 567–576. [Google Scholar] [CrossRef]
- Marsili, A.; Tang, D.; Harney, J.; Singh, P.; Zavacki, A.; Dentice, M.; Salvatore, D.; Larsen, P. Type II iodothyronine deiodinase provides intracellular 3,5,3’-triiodothyronine to normal and regenerating mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E818–E824. [Google Scholar] [CrossRef] [PubMed]
- Iwen, K.; Oelkrug, R.; Kalscheuer, H.; Brabant, G. Metabolic Syndrome in Thyroid Disease. Front. Horm. Res. 2018, 49, 48–66. [Google Scholar]
- Mabilleau, G.; Pereira, M.; Chenu, C. Novel skeletal effects of glucagon-like peptide-1 (GLP-1) receptor agonists. J. Endocrinol. 2018, 236, R29–R42. [Google Scholar] [CrossRef]
- Galton, V.A. The roles of the iodothyronine deiodinases in mammalian development. Thyroid 2005, 15, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.A.; Dorfman, D.M.; Genest, D.R.; Salvatore, D.; Larsen, P.R. Type 3 iodothyronine deiodinase is highly expressed in the human uteroplacental unit and in fetal epithelium. J. Clin. Endocrinol. Metab. 2003, 88, 1384–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuer, H. The importance of thyroid hormone transporters for brain development and function. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 265–276. [Google Scholar] [CrossRef]
- Bernal, J.; Guadano-Ferraz, A.; Morte, B. Thyroid hormone transporters-functions and clinical implications. Nat. Rev. Endocrinol. 2015, 11, 690. [Google Scholar] [CrossRef] [Green Version]
- Joffe, R.; Sokolov, S. Thyroid hormones, the brain, and affective disorders. Crit. Rev. Neurobiol. 1994, 8, 45–63. [Google Scholar]
- Galton, V.A.; Wood, E.T.; Germain, E.A.S.; Withrow, C.-A.; Aldrich, G.; Germain, G.M.S.; Clark, A.S.; Germain, D.L.S. Thyroid Hormone Homeostasis and Action in the Type 2 Deiodinase-Deficient Rodent Brain during Development. Endocrinology 2007, 148, 3080–3088. [Google Scholar] [CrossRef]
- Galton, V.; Schneider, M.; Clark, A.; Germain, D.S. Life without thyroxine to 3,5,3’-triiodothyronine conversion: Studies in mice devoid of the 5’-deiodinases. Endocrinology 2009, 150, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Bárez-López, S.; Bosch-García, D.; Gómez-Andrés, D.; Pulido-Valdeolivas, I.; Montero-Pedrazuela, A.; Obregon, M.J.; Guadaño-Ferraz, A. Abnormal Motor Phenotype at Adult Stages in Mice Lacking Type 2 Deiodinase. PLoS ONE 2014, 9, e103857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bárez-López, S.; Montero-Pedrazuela, A.; Bosch-García, D.; Venero, C.; Guadaño-Ferraz, A. Increased anxiety and fear memory in adult mice lacking type 2 deiodinase. Psychoneuroendocrinology 2017, 84, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Fonseca, T.L.; Bocco, B.M.L.C.; Fernandes, G.W.; McAninch, E.A.; Bolin, A.P.; Da Conceição, R.R.; Werneck-De-Castro, J.P.; Ignacio, D.L.; Egri, P.; et al. Type 2 deiodinase polymorphism causes ER stress and hypothyroidism in the brain. J. Clin. Investig. 2018, 129, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Aguilera, N.; Thévenet, M.; Dkhissi-Benyahya, O.; Flamant, F. Neuronal expression of a thyroid hormone receptor α mutation alters mouse behaviour. Behav. Brain Res. 2017, 321, 18–27. [Google Scholar] [CrossRef]
- Heinrich, T.; Grahm, G. Hypothyroidism Presenting as Psychosis: Myxedema Madness Revisited. Prim. Care Companion J. Clin. Psychiatry 2003, 5, 260–266. [Google Scholar] [CrossRef]
- Gönen, M.S.; Kisakol, G.; Cilli, A.S.; Dikbas, O.; Gungor, K.; Inal, A.; Kaya, A. Assessment of anxiety in subclinical thyroid disorders. Endocr. J. 2004, 51, 311–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.; Goetz, T.; Glenn, T.; Whybrow, P.C. The thyroid-brain interaction in thyroid disorders and mood disorders. J. Neuroendocr. 2008, 20, 1101–1114. [Google Scholar] [CrossRef]
- De Luca, R.; Davis, P.J.; Lin, H.-Y.; Gionfra, F.; Percario, Z.A.; Affabris, E.; Pedersen, J.Z.; Marchese, C.; Trivedi, P.; Anastasiadou, E.; et al. Thyroid Hormones Interaction With Immune Response, Inflammation and Non-thyroidal Illness Syndrome. Front. Cell Dev. Biol. 2021, 8, 614030. [Google Scholar] [CrossRef]
- van der Spek, A.; Fliers, E.; Boelen, A. Thyroid Hormone and Deiodination in Innate Immune Cells. Endocrinology 2021, 162, bqaa200. [Google Scholar] [CrossRef]
- Bernal, J. Thyroid Hormones in Brain Development and Function; Feingold, K., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W., Dhatariya, K., Dungan, K., Hershman, J., Hofland, J., Kalra, S., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Lemkine, G.F.; Raji, A.; Alfama, G.; Turque, N.; Hassani, Z.; Alegria-Prévot, O.; Samarut, J.; Levi, G.; Demeneix, B.A. Adult neural stem cell cycling in vivo requires thyroid hormone and its alpha receptor. FASEB J. 2005, 19, 863–865. [Google Scholar] [CrossRef] [Green Version]
- Suda, S.; Muraga, K.; Kanamaru, T.; Okubo, S.; Abe, A.; Aoki, J.; Suzuki, K.; Sakamoto, Y.; Shimoyama, T.; Nito, C.; et al. Low free triiodothyronine predicts poor functional outcome after acute ischemic stroke. J. Neurol. Sci. 2016, 368, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Mendes-De-Aguiar, C.B.N.; Alchini, R.; Decker, H.; Alvarez-Silva, M.; Tasca, C.I.; Trentin, A.G. Thyroid hormone increases astrocytic glutamate uptake and protects astrocytes and neurons against glutamate toxicity. J. Neurosci. Res. 2008, 86, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Rami, A.; Krieglstein, J. Thyroxine attenuates hippocampal neuronal damage caused by ischemia in the rat. Life Sci. 1992, 50, 645–650. [Google Scholar] [CrossRef]
- Genovese, T.; Impellizzeri, D.; Ahmad, A.; Cornelius, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E. Post-ischaemic thyroid hormone treatment in a rat model of acute stroke. Brain Res. 2013, 1513, 92–102. [Google Scholar] [CrossRef]
- Li, J.; Donangelo, I.; Abe, K.; Scremin, O.; Ke, S.; Li, F.; Milanesi, A.; Liu, Y.-Y.; Brent, G.A. Thyroid hormone treatment activates protective pathways in both in vivo and in vitro models of neuronal injury. Mol. Cell. Endocrinol. 2017, 452, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, T.; Akbari, M.; Malek, F.; Kashani, I.R.; Rastegar, T.; Noorbakhsh, F.; Ghazi-Khansari, M.; Attari, F.; Hassanzadeh, G. Improvement of memory and learning by intracerebroventricular microinjection of T3 in rat model of ischemic brain stroke mediated by upregulation of BDNF and GDNF in CA1 hippocampal region. DARU J. Pharm. Sci. 2017, 25, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boltzmann, M.; Schmidt, S.B.; Rollnik, J.D. Impact of Thyroid Hormone Levels on Functional Outcome in Neurological and Neurosurgical Early Rehabilitation Patients. BioMed Res. Int. 2017, 2017, 4719279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourbopoulos, A.; Mourouzis, I.; Karapanayiotides, T.; Nousiopoulou, E.; Chatzigeorgiou, S.; Mavridis, T.; Kokkinakis, I.; Touloumi, O.; Irinopoulou, T.; Chouliaras, K.; et al. Changes in thyroid hormone receptors after permanent cerebral ischemia in male rats. J. Mol. Neurosci. 2014, 54, 78–91. [Google Scholar] [CrossRef]
- Margaill, I.; Royer, J.; Lerouet, D.; Ramaugé, M.; Le Goascogne, C.; Li, W.W.; Plotkine, M.; Pierre, M.; Courtin, F. Induction of Type 2 iodothyronine deiodinase in astrocytes after transient focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 2005, 25, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Przybylak, M.; Grabowski, J.; Bidzan, L. Cognitive functions and thyroid hormones secretion disorders. Psychiatr. Polska 2021, 55, 309–321. [Google Scholar] [CrossRef]
- Delpont, B.; Aboa-Eboulé, C.; Durier, J.; Petit, J.-M.; Daumas, A.; Legris, N.; Daubail, B.; Giroud, M.; Béjot, Y. Associations between Thyroid Stimulating Hormone Levels and Both Severity and Early Outcome of Patients with Ischemic Stroke. Eur. Neurol. 2016, 76, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, D.; Xiong, Y.; Yuan, R.; Tao, W.; Liu, M. Low free triiodothyronine levels are related to symptomatic intracranial hemorrhage and poor functional outcomes after intravenous thrombolysis in acute ischemic stroke patients. Neurol. Res. 2016, 38, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Lamba, N.; Liu, C.; Zaidi, H.; Broekman, M.; Simjian, T.; Shi, C.; Doucette, J.; Ren, S.; Smith, T.R.; Mekary, R.A.; et al. A prognostic role for Low tri-iodothyronine syndrome in acute stroke patients: A systematic review and meta-analysis. Clin. Neurol. Neurosurg. 2018, 169, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.I.; Suri, F.K.; Nasar, A.; Kirmani, J.F.; Divani, A.A.; Giles, W.H. Free thyroxine index and risk of stroke: Results from the National Health and Nutrition Examination Survey Follow-up Study. J. Pharmacol. Exp. Ther. 2006, 12, CR501–CR506. [Google Scholar]
- Dhital, R.; Poudel, D.R.; Tachamo, N.; Gyawali, B.; Basnet, S.; Shrestha, P.; Karmacharya, P. Ischemic Stroke and Impact of Thyroid Profile at Presentation: A Systematic Review and Meta-analysis of Observational Studies. J. Stroke Cerebrovasc. Dis. 2017, 26, 2926–2934. [Google Scholar] [CrossRef]
- Jiang, X.; Xingjun, J.; Wu, J.; Du, R.; Liu, H.; Chen, J.; Wang, J.; Wang, C.; Wu, Y. Prognostic value of thyroid hormones in acute ischemic stroke—A meta analysis. Sci. Rep. 2017, 7, 16256. [Google Scholar] [CrossRef] [Green Version]
- Hama, S.; Kitaoka, T.; Shigenobu, M.; Watanabe, A.; Imura, I.; Seno, H.; Tominaga, A.; Arita, K.; Kurisu, K. Malnutrition and nonthyroidal illness syndrome after stroke. Metabolism 2005, 54, 699–704. [Google Scholar] [CrossRef]
- Ambrosius, W.; Kazmierski, R.; Gupta, V.; Warot, A.W.; Adamczewska-Kociałkowska, D.; Błazejewska, A.; Ziemnicka, K.; Nowinski, W.L. Low Free Triiodothyronine Levels are Related to Poor Prognosis in Acute Ischemic Stroke. Exp. Clin. Endocrinol. Diabetes 2010, 119, 139–143. [Google Scholar] [CrossRef]
- Zhang, Y.; Meyer, M.A. Clinical analysis on alteration of thyroid hormones in the serum of patients with acute ischemic stroke. Stroke Res. Treat. 2010, 2010, 290678. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.-Y.; Li, W.-Y.; Hu, X.-Y. Alteration of Thyroid-Related Hormones within Normal Ranges and Early Functional Outcomes in Patients with Acute Ischemic Stroke. Int. J. Endocrinol. 2016, 2016, 3470490. [Google Scholar] [CrossRef] [Green Version]
- Chaker, L.; Baumgartner, C.; Elzen, W.P.J.D.; Collet, T.-H.; Ikram, M.A.; Blum, M.R.; Dehghan, A.; Drechsler, C.; Luben, R.N.; Portegies, M.L.P.; et al. Thyroid Function Within the Reference Range and the Risk of Stroke: An Individual Participant Data Analysis. J. Clin. Endocrinol. Metab. 2016, 101, 4270–4282. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, L.; Godbole, M.M.; Sinha, R.A.; Pradhan, S. Reverse triiodothyronine (rT3) attenuates ischemia-reperfusion injury. Biochem. Biophys. Res. Commun. 2018, 506, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, S.; Dehghani, G.A. Cerebral Ischemia/Reperfusion Injury in the Hyperthyroid Rat. Iran. J. Med. Sci. 2017, 42, 48–56. [Google Scholar] [PubMed]
- Yang, M.-H.; Yang, F.-Y.; Lee, D.-D. Thyroid disease as a risk factor for cerebrovascular disease. J. Stroke Cerebrovasc. Dis. 2015, 24, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Aslan, M.; Cosar, N.; Celik, H.; Aksoy, N.; Dulger, A.C.; Begenik, H.; Soyoral, Y.U.; Kucukoglu, M.E.; Selek, S. Evaluation of oxidative status in patients with hyperthyroidism. Endocrine 2011, 40, 285–289. [Google Scholar] [CrossRef]
- Franklyn, J.; Maisonneuve, P.; Sheppard, M.; Betteridge, J.; Boyle, P. Mortality after the treatment of hyperthyroidism with radioactive iodine. N. Engl. J. Med. 1998, 338, 712–718. [Google Scholar] [CrossRef]
- Brüssel, T.; Matthay, M.A.; Chernow, B. Pulmonary manifestations of endocrine and metabolic disorders. Clin. Chest Med. 1989, 10, 645–653. [Google Scholar] [CrossRef]
- Alevizaki, M.; Synetou, M.; Xynos, K.; Alevizaki, C.C.; Vemmos, K.N. Hypothyroidism as a protective factor in acute stroke patients. Clin. Endocrinol. 2006, 65, 369–372. [Google Scholar] [CrossRef]
- Remmel, K.S.; Wanahita, A.; Moore, K.; Gruenthal, M. Acute ischemic stroke and hypothyroidism. J. Ky. Med. Assoc. 2006, 104, 191–193. [Google Scholar]
- Pantos, C.; Mourouzis, C.; Katramadou, M.; Saranteas, T.; Mourouzis, I.; Karageorgiou, H.; Tesseromatis, C.; Kostopanagiotou, G.; Asimacopoulos, P.; Cokkinos, D.V. Decreased vascular reactivity to alpha1 adrenergic stimulation in the presence of hypothyroid state: A part of an adaptive response? Int. Angiol. 2006, 25, 216–220. [Google Scholar]
- Talhada, D.; Feiteiro, J.; Costa, A.R.; Talhada, T.; Cairrão, E.; Wieloch, T.; Englund, E.; Santos, C.R.; Gonçalves, I.; Ruscher, K. Triiodothyronine modulates neuronal plasticity mechanisms to enhance functional outcome after stroke. Acta Neuropathol. Commun. 2019, 7, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luitse, M.J.; van Seeters, T.; Horsch, A.D.; Kool, H.A.; Velthuis, B.K.; Kappelle, L.J.; Biessels, G.J. Admission hyperglycaemia and cerebral perfusion deficits in acute ischaemic stroke. Cerebrovasc. Dis. 2013, 35, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Bashyal, S.; Gorkhaly, M.P.; Devkota, R.; Devkota, R.; Regmi, P.R.; Amatya, I. Alteration of Thyroid Hormone among Patients with Ischemic Stroke visiting a Tertiary Care Hospital: A Descriptive Cross-sectional Study. J. Nepal Med. Assoc. 2021, 59, 779–782. [Google Scholar] [CrossRef]
- Bunevicius, A.; Iervasi, G.; Bunevicius, R. Neuroprotective actions of thyroid hormones and low-T3 syndrome as a biomarker in acute cerebrovascular disorders. Expert Rev. Neurother. 2015, 15, 315–326. [Google Scholar] [CrossRef]
- Suda, S.; Aoki, J.; Shimoyama, T.; Suzuki, K.; Sakamoto, Y.; Katano, T.; Okubo, S.; Nito, C.; Nishiyama, Y.; Mishina, M.; et al. Low Free Triiodothyronine at Admission Predicts Poststroke Infection. J. Stroke Cerebrovasc. Dis. 2017, 27, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yang, C.; Wang, H. Free Triiodothyronine Is Associated with Poor Outcomes after Acute Ischemic Stroke. Int. J. Clin. Pract. 2022, 2022, 1982193. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Fang, M.; Liu, X. Low free triiodothyronine levels predict symptomatic intracranial hemorrhage and worse short-term outcome of thrombolysis in patients with acute ischemia stroke. Medicine 2017, 96, e8539. [Google Scholar] [CrossRef]
- Chen, H.; Wu, Y.; Huang, G.; He, W.; Lin, S.; Zhang, X.; He, J. Low Tri-iodothyronine Syndrome Is Associated With Cognitive Impairment in Patients With Acute Ischemic Stroke: A Prospective Cohort Study. Am. J. Geriatr. Psychiatry 2018, 26, 1222–1230. [Google Scholar] [CrossRef]
- Chen, H.; Xu, M.; Huang, Y.; He, J.; Ren, W. Low triiodothyronine syndrome is associated with stroke-associated pneumonia. Eur. J. Clin. Investig. 2022, 52, e13840. [Google Scholar] [CrossRef]
- Czap, A.; Shoup, J.P.; Winkler, J.; Staff, I.; Fortunato, G.; Malchoff, C.; McCullough, L.D.; Sansing, L.H. Intracerebral hemorrhage with hypothyroidism. J. Stroke Cerebrovasc. Dis. 2013, 22, e602–e609. [Google Scholar] [CrossRef]
- Kim, H.J.; Kang, T.; Kang, M.J.; Ahn, H.S.; Sohn, S.Y. Incidence and Mortality of Myocardial Infarction and Stroke in Patients with Hyperthyroidism: A Nationwide Cohort Study in Korea. Thyroid 2020, 30, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Papaleontiou, M.; Levine, D.A.; Reyes-Gastelum, D.; Hawley, S.T.; Banerjee, M.; Haymart, M.R. Thyroid Hormone Therapy and Incident Stroke. J. Clin. Endocrinol. Metab. 2021, 106, e3890–e3900. [Google Scholar] [CrossRef] [PubMed]
- Wollenweber, F.A.; Zietemann, V.; Gschwendtner, A.; Opherk, C.; Dichgans, M. Subclinical hyperthyroidism is a risk factor for poor functional outcome after ischemic stroke. Stroke 2013, 44, 1446–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fani, L.; Dueñas, O.R.; Bos, D.; Vernooij, M.W.; Klaver, C.C.W.; Ikram, M.K.; Peeters, R.P.; Chaker, L. Thyroid Status and Brain Circulation: The Rotterdam Study. J. Clin. Endocrinol. Metab. 2021, 107, e1293–e1302. [Google Scholar] [CrossRef]
- Chen, Z.; Sun, Y.; Zhang, Y.; He, Y.; Chen, H.; Su, Y. Low TSH level predicts a poor clinical outcome in patients with anterior circulation ischemic stroke after endovascular thrombectomy. Neurol. Sci. 2020, 41, 1821–1828. [Google Scholar] [CrossRef]
- Wang, J.; Li, F.; Xiao, L.; Peng, F.; Sun, W.; Li, M.; Liu, D.; Jiang, Y.; Guo, R.; Li, H.; et al. Depressed TSH level as a predictor of poststroke fatigue in patients with acute ischemic stroke. Neurology 2018, 91, e1971–e1978. [Google Scholar] [CrossRef]
- Møllehave, L.T.; Skaaby, T.; Linneberg, A.; Knudsen, N.; Jørgensen, T.; Thuesen, B.H. The association of thyroid stimulation hormone levels with incident ischemic heart disease, incident stroke, and all-cause mortality. Endocrine 2020, 68, 358–367. [Google Scholar] [CrossRef]
- Guo, J.; Wang, J.; Xia, Y.; Jiang, S.; Xu, P.; Tao, C.; Sun, W.; Liu, X. Thyroid Function Affects the Risk of Post-stroke Depression in Patients With Acute Lacunar Stroke. Front. Neurol. 2022, 13, 792843. [Google Scholar] [CrossRef]
- Bogush, N.; Tan, L.; Naqvi, E.; Calvert, J.; Graham, R.; Taylor, W.; Naqvi, N.; Husain, A. Remuscularization with triiodothyronine and beta1-blocker therapy reverses post-ischemic left ventricular dysfunction and adverse remodeling. Sci. Rep. 2022, 12, 8852. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef]
- Chattergoon, N.N.; Giraud, G.D.; Louey, S.; Stork, P.; Fowden, A.L.; Thornburg, K.L. Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J. 2011, 26, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschou, S.A.; Bletsa, E.; Stampouloglou, P.K.; Tsigkou, V.; Valatsou, A.; Stefanaki, K.; Kazakou, P.; Spartalis, M.; Spartalis, E.; Oikonomou, E.; et al. Thyroid disorders and cardiovascular manifestations: An update. Endocrine 2022, 75, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.; Huang, G.N. Endocrine Influence on Cardiac Metabolism in Development and Regeneration. Endocrinology 2021, 162, bqab081. [Google Scholar] [CrossRef] [PubMed]
- Cicatiello, A.; Sagliocchi, S.; Nappi, A.; Di Cicco, E.; Miro, C.; Murolo, M.; Stornaiuolo, M.; Dentice, M. Thyroid hormone regulates glutamine metabolism and anaplerotic fluxes by inducing mitochondrial glutamate aminotransferase GPT2. Cell Rep. 2022, 38, 110562. [Google Scholar] [CrossRef]
- Simkó, J.; Barta, K.; Szabó, Z.; Varga, E.; Nagy, E.; Lorincz, I. Cardiovascular manifestations of thyrotoxicosis and thyroid dysfunction caused by amiodarone. Orvosi Hetil. 2004, 145, 2411–2417. [Google Scholar]
- Hyyti, O.M.; Portman, M.A. Molecular mechanisms of cross-talk between thyroid hormone and peroxisome proliferator activated receptors: Focus on the heart. Cardiovasc. Drugs Ther. 2006, 20, 463–469. [Google Scholar] [CrossRef]
- von Hafe, M.; Neves, J.S.; Vale, C.; Borges-Canha, M.; Leite-Moreira, A. The impact of thyroid hormone dysfunction on ischemic heart disease. Endocr. Connect. 2019, 8, R76–R90. [Google Scholar] [CrossRef]
- Gupta, M.P. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2007, 43, 388–403. [Google Scholar] [CrossRef] [Green Version]
- Zuurbier, C.; Bertrand, L.; Beauloye, C.; Andreadou, I.; Ruiz-Meana, M.; Jespersen, N.; Kula-Alwar, D.; Prag, H.; Botker, H.E.; Dambrova, M.; et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J. Cell Mol. Med. 2020, 24, 5937–5954. [Google Scholar] [CrossRef]
- Lee, E.C.; Ha, T.W.; Lee, D.-H.; Hong, D.-Y.; Park, S.-W.; Lee, J.Y.; Lee, M.R.; Oh, J.S. Utility of Exosomes in Ischemic and Hemorrhagic Stroke Diagnosis and Treatment. Int. J. Mol. Sci. 2022, 23, 8367. [Google Scholar] [CrossRef]
- Razvi, S.; Jabbar, A.; Pingitore, A.; Danzi, S.; Biondi, B.; Klein, I.; Peeters, R.; Zaman, A.; Iervasi, G. Thyroid Hormones and Cardiovascular Function and Diseases. J. Am. Coll. Cardiol. 2018, 71, 1781–1796. [Google Scholar] [CrossRef] [PubMed]
- Sagliocchi, S.; Cicatiello, A.G.; Di Cicco, E.; Ambrosio, R.; Miro, C.; Di Girolamo, D.; Nappi, A.; Mancino, G.; De Stefano, M.A.; Luongo, C.; et al. The thyroid hormone activating enzyme, type 2 deiodinase, induces myogenic differentiation by regulating mitochondrial metabolism and reducing oxidative stress. Redox Biol. 2019, 24, 101228. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Liu, L.; Liao, X.; Zhang, C.; Ruan, H. Thyroid hormone protects cardiomyocytes from H2O2-induced oxidative stress via the PI3K-AKT signaling pathway. Exp. Cell Res. 2019, 380, 205–215. [Google Scholar] [CrossRef]
- Baumgartner, C.; da Costa, B.R.; Collet, T.-H.; Feller, M.; Floriani, C.; Bauer, D.C.; Cappola, A.R.; Heckbert, S.R.; Ceresini, G.; Gussekloo, J.; et al. Thyroid Function Within the Normal Range, Subclinical Hypothyroidism, and the Risk of Atrial Fibrillation. Circulation 2017, 136, 2100–2116. [Google Scholar] [CrossRef]
- Sawin, C.T.; Geller, A.; Wolf, P.A.; Belanger, A.J.; Baker, E.; Bacharach, P.; Wilson, P.; Benjamin, E.J.; D’Agostino, R.B. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N. Engl. J. Med. 1994, 331, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Selmer, C.; Olesen, J.B.; Hansen, M.L.; Lindhardsen, J.; Olsen, A.-M.S.; Madsen, J.C.; Faber, J.; Hansen, P.R.; Pedersen, O.D.; Torp-Pedersen, C.; et al. The spectrum of thyroid disease and risk of new onset atrial fibrillation: A large population cohort study. BMJ 2012, 345, e7895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammage, M.; Parle, J.; Holder, R.; Roberts, L.; Hobbs, F.; Wilson, S.; Sheppard, M.; Franklyn, J. Association between serum free thyroxine concentration and atrial fibrillation. Arch. Intern. Med. 2007, 167, 928–934. [Google Scholar] [CrossRef]
- Cappola, A.R.; Ladenson, P.W. Hypothyroidism and atherosclerosis. J. Clin. Endocrinol. Metab. 2003, 88, 2438–2444. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.; Taha, W.; Kundumadam, S.; Khan, M. Atrial fibrillation and hyperthyroidism: A literature review. Indian Heart J. 2017, 69, 545–550. [Google Scholar] [CrossRef]
- Larsson, S.; Allara, E.; Mason, A.; Michaelsson, K.; Burgess, S. Thyroid Function and Dysfunction in Relation to 16 Cardiovascular Diseases. Circ. Genom Precis Med. 2019, 12, e002468. [Google Scholar] [CrossRef] [Green Version]
- Iervasi, G.; Pingitore, A.; Landi, P.; Raciti, M.; Ripoli, A.; Scarlattini, M.; L’Abbate, A.; Donato, L. Low-T3 syndrome: A strong prognostic predictor of death in patients with heart disease. Circulation 2003, 107, 708–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lymvaios, I.; Mourouzis, I.; Cokkinos, D.V.; Dimopoulos, M.A.; Toumanidis, S.T.; Pantos, C. Thyroid hormone and recovery of cardiac function in patients with acute myocardial infarction: A strong association? Eur. J. Endocrinol. 2011, 165, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbar, A.; Pingitore, A.; Pearce, S.H.S.; Zaman, A.; Iervasi, G.; Razvi, S. Thyroid hormones and cardiovascular disease. Nat. Rev. Cardiol. 2017, 14, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Novitzky, D.; Cooper, D.K.C. Thyroid hormone and the stunned myocardium. J. Endocrinol. 2014, 223, R1–R8. [Google Scholar] [CrossRef] [Green Version]
- Eagan, D.; Spencer-Bonilla, G.; Maraka, S.; Aggarwal, M.; Ospina, N.S. Management of Hypothyroidism in Patients with Acute Myocardial Infarction. Medicina 2020, 56, 214. [Google Scholar] [CrossRef]
- Danzi, S.; Klein, I. Thyroid Abnormalities in Heart Failure. Heart Fail. Clin. 2019, 16, 1–9. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murolo, M.; Di Vincenzo, O.; Cicatiello, A.G.; Scalfi, L.; Dentice, M. Cardiovascular and Neuronal Consequences of Thyroid Hormones Alterations in the Ischemic Stroke. Metabolites 2023, 13, 22. https://doi.org/10.3390/metabo13010022
Murolo M, Di Vincenzo O, Cicatiello AG, Scalfi L, Dentice M. Cardiovascular and Neuronal Consequences of Thyroid Hormones Alterations in the Ischemic Stroke. Metabolites. 2023; 13(1):22. https://doi.org/10.3390/metabo13010022
Chicago/Turabian StyleMurolo, Melania, Olivia Di Vincenzo, Annunziata Gaetana Cicatiello, Luca Scalfi, and Monica Dentice. 2023. "Cardiovascular and Neuronal Consequences of Thyroid Hormones Alterations in the Ischemic Stroke" Metabolites 13, no. 1: 22. https://doi.org/10.3390/metabo13010022