
Divergent Thyroid Hormone Levels in Plasma and Left Ventricle of the Heart in Compensated and Decompensated Cardiac Hypertrophy Induced by Chronic Adrenergic Stimulation in Mice
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Isoproterenol Induces Compensated Left-Ventricular Hypertrophy in cD3KO-CS Mice
2.1.1. Cardiac Parameters of ISO-Infused cD3KO-CS Mice
2.1.2. Thyroid Hormone Levels and Type 3 Deiodinase Activity in ISO-Infused cD3KO-CS Mice
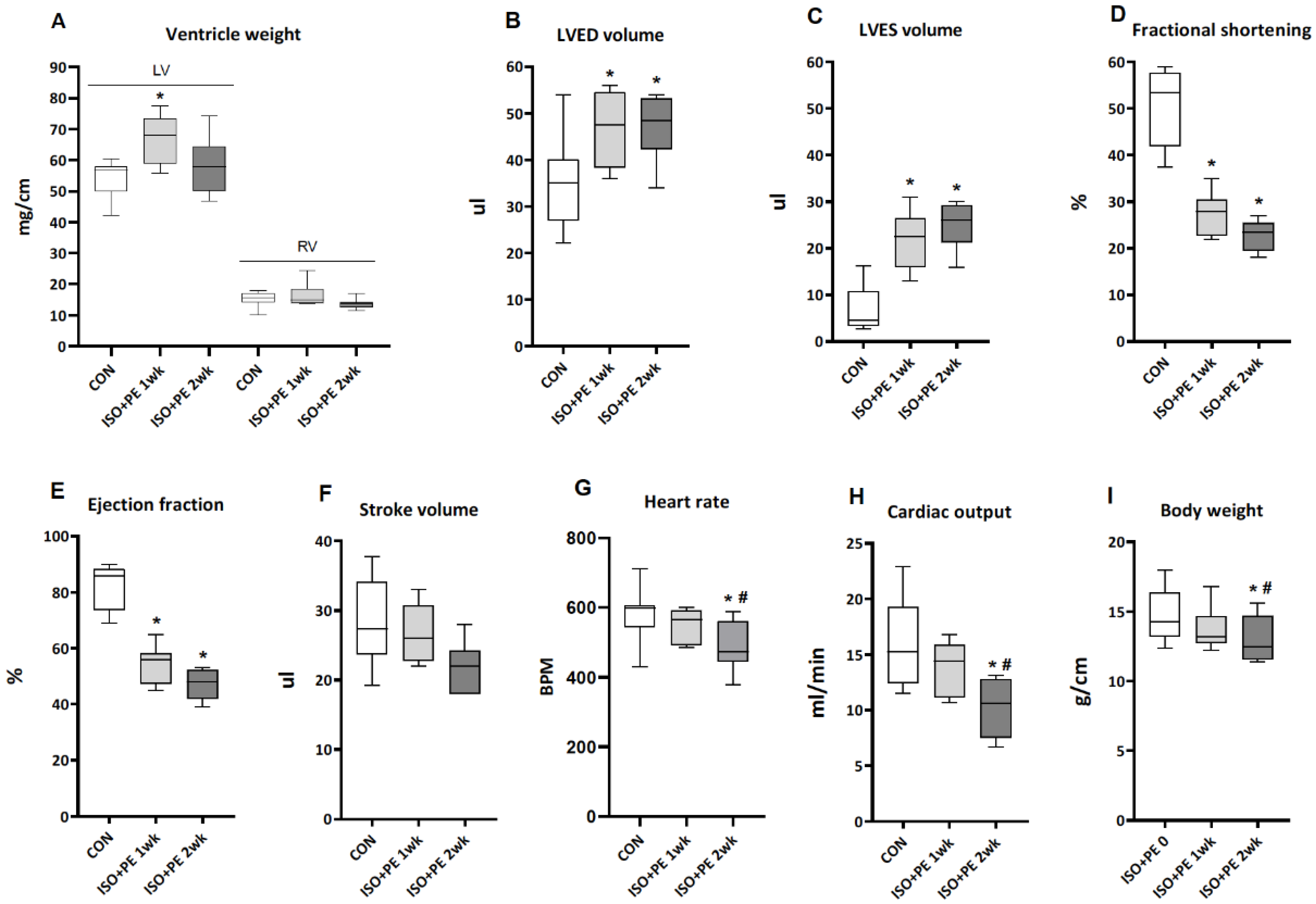
2.2. Isoproterenol + Phenylephrine Induces Decompensated Left-Ventricular Hypertrophy in cD3KO-CS Mice
2.2.1. Cardiac Parameters of ISO + PE-Infused cD3KO-CS Mice
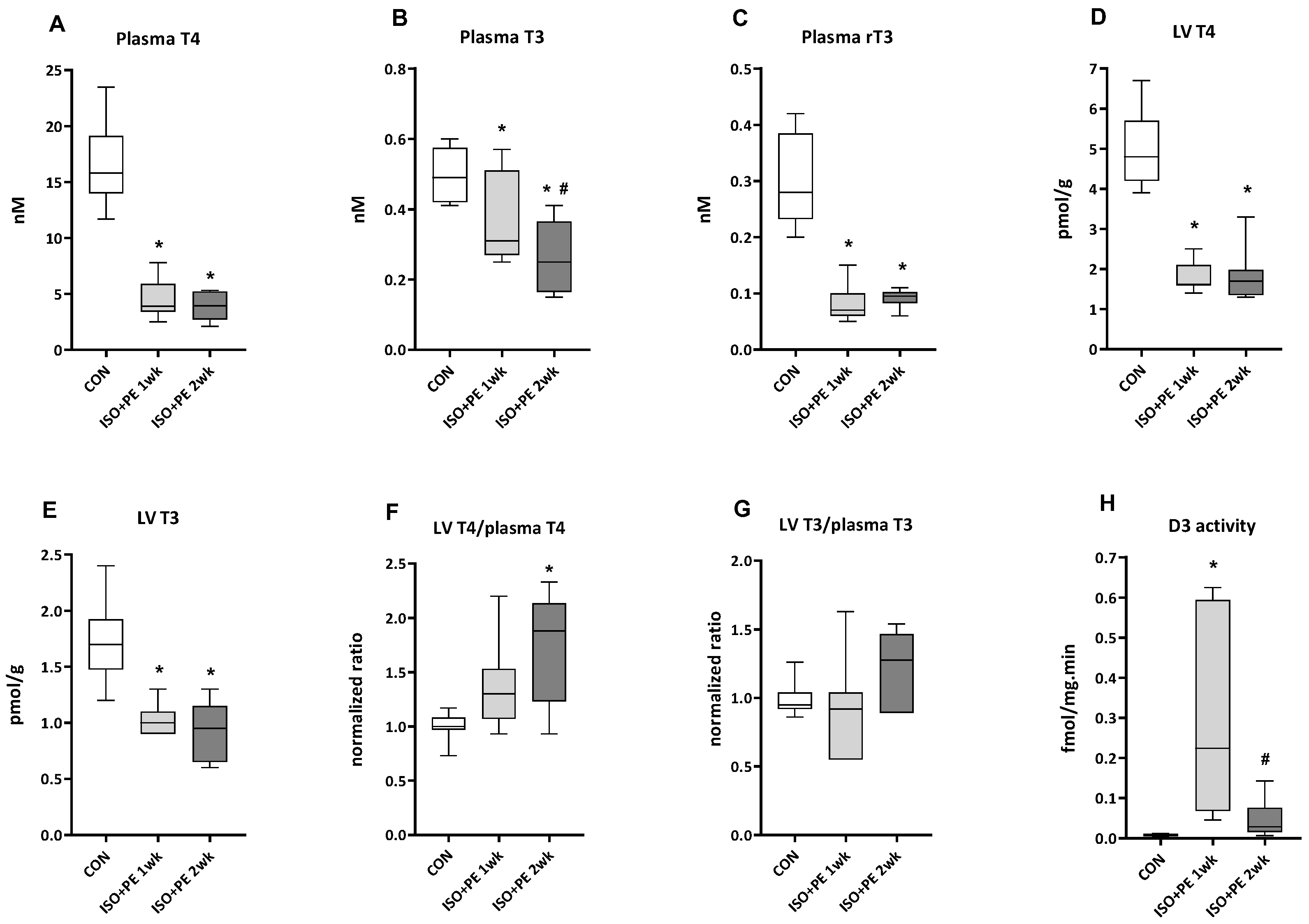
2.2.2. Thyroid Hormone Levels and Type 3 Deiodinase Activity in ISO + PE-Infused cD3KO-CS Mice
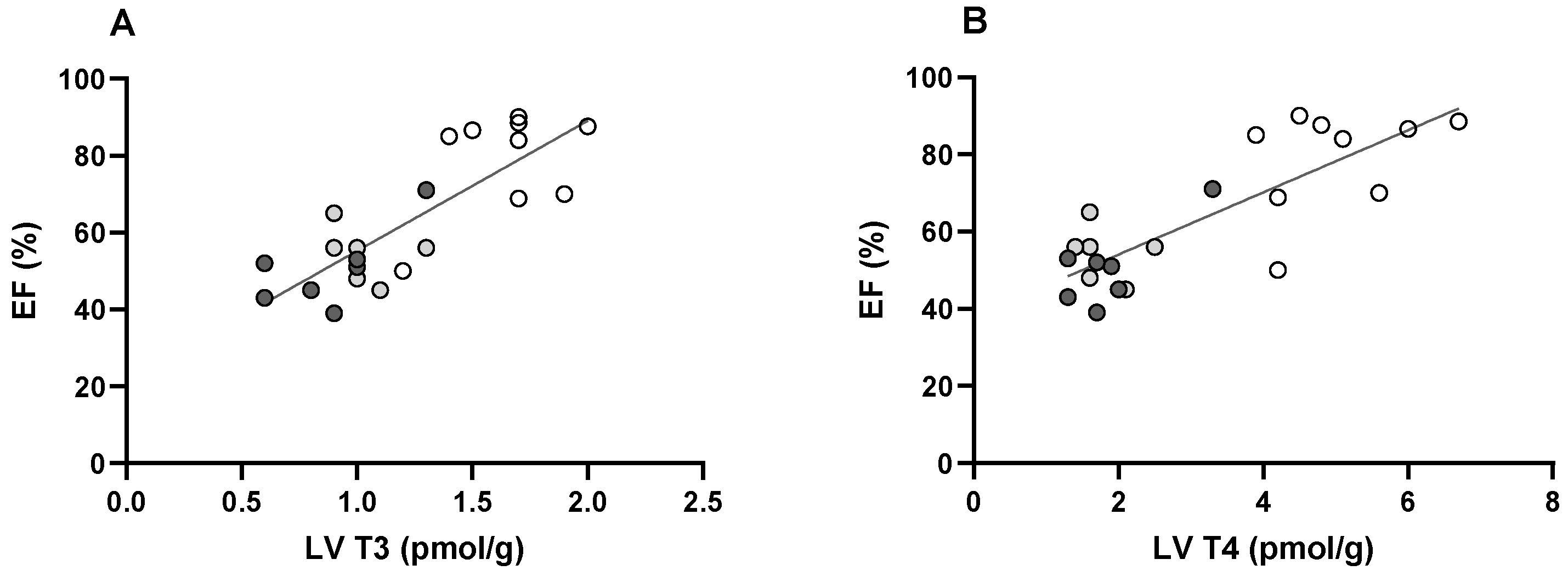
3. Discussion
4. Materials and Methods
4.1. Generation of the cD3KO-CS Strain
4.2. Implantation of Osmotic Minipumps
4.3. Echocardiography
4.4. Tissue Collection
4.5. Determination of TH Levels in LV Tissue and Plasma
4.6. Determination of D3 Activity
4.7. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular Distinction between Physiological and Pathological Cardiac Hypertrophy: Experimental Findings and Therapeutic Strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef] [PubMed]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular Basis of Physiological Heart Growth: Fundamental Concepts and New Players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heineke, J.; Molkentin, J.D. Regulation of Cardiac Hypertrophy by Intracellular Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative Stress as a Mechanism for Functional Alterations in Cardiac Hypertrophy and Heart Failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef]
- Hönes, G.S.; Rakov, H.; Logan, J.; Liao, X.-H.; Werbenko, E.; Pollard, A.S.; Præstholm, S.M.; Siersbæk, M.S.; Rijntjes, E.; Gassen, J.; et al. Noncanonical Thyroid Hormone Signaling Mediates Cardiometabolic Effects in Vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E11323–E11332. [Google Scholar] [CrossRef] [Green Version]
- Klein, I.; Ojamaa, K. Thyroid Hormone and the Cardiovascular System. N. Engl. J. Med. 2001, 344, 501–509. [Google Scholar] [CrossRef]
- Jabbar, A.; Pingitore, A.; Pearce, S.H.S.; Zaman, A.; Iervasi, G.; Razvi, S. Thyroid Hormones and Cardiovascular Disease. Nat. Rev. Cardiol. 2016, 14, 39–55. [Google Scholar] [CrossRef]
- Cokkinos, D.V.; Chryssanthopoulos, S. Thyroid Hormones and Cardiac Remodeling. Heart Fail. Rev. 2016, 21, 365–372. [Google Scholar] [CrossRef]
- Danzi, S.; Klein, I. Thyroid Disease and the Cardiovascular System. Endocrinol. Metab. Clin. N. Am. 2014, 43, 517–528. [Google Scholar] [CrossRef]
- Gerdes, A.M. Restoration of Thyroid Hormone Balance: A Game Changer in the Treatment of Heart Failure? AJP Heart Circ. Physiol. 2015, 308, H1–H10. [Google Scholar] [CrossRef] [Green Version]
- Fliers, E.; Boelen, A. An Update on Non-Thyroidal Illness Syndrome. J. Endocrinol. Investig. 2021, 44, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Wassen, F.W.J.S.; Schiel, A.E.; Kuiper, G.G.J.M.; Kaptein, E.; Bakker, O.; Visser, T.J.; Simonides, W.S. Induction of Thyroid Hormone-Degrading Deiodinase in Cardiac Hypertrophy and Failure. Endocrinology 2002, 143, 2812–2815. [Google Scholar] [CrossRef] [PubMed]
- Simonides, W.S.; Mulcahey, M.A.; Redout, E.M.; Muller, A.; Zuidwijk, M.J.; Visser, T.J.; Wassen, F.W.J.S.; Crescenzi, A.; Da-Silva, W.S.; Harney, J.; et al. Hypoxia-Inducible Factor Induces Local Thyroid Hormone Inactivation during Hypoxic-Ischemic Disease in Rats. J. Clin. Investig. 2008, 118, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Pol, C.J.; Muller, A.; Zuidwijk, M.J.; van Deel, E.D.; Kaptein, E.; Saba, A.; Marchini, M.; Zucchi, R.; Visser, T.J.; Paulus, W.J.; et al. Left-Ventricular Remodeling After Myocardial Infarction Is Associated with a Cardiomyocyte-Specific Hypothyroid Condition. Endocrinology 2011, 152, 669–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivieri, M.G.; Oudit, G.Y.; Sah, R.; Kerfant, B.-G.; Sun, H.; Gramolini, A.O.; Pan, Y.; Wickenden, A.D.; Croteau, W.; Morreale de Escobar, G.; et al. Cardiac-Specific Elevations in Thyroid Hormone Enhance Contractility and Prevent Pressure Overload-Induced Cardiac Dysfunction. Proc. Natl. Acad. Sci. USA 2006, 103, 6043–6048. [Google Scholar] [CrossRef] [Green Version]
- Olivares, E.L.; Marassi, M.P.; Fortunato, R.S.; da Silva, A.C.M.; Costa-e-Sousa, R.H.; Araújo, I.G.; Mattos, E.C.; Masuda, M.O.; Mulcahey, M.A.; Huang, S.A.; et al. Thyroid Function Disturbance and Type 3 Iodothyronine Deiodinase Induction after Myocardial Infarction in Rats a Time Course Study. Endocrinology 2007, 148, 4786–4792. [Google Scholar] [CrossRef] [Green Version]
- Ueta, C.B.; Oskouei, B.N.; Olivares, E.L.; Pinto, J.R.; Correa, M.M.; Simovic, G.; Simonides, W.S.; Hare, J.M.; Bianco, A.C. Absence of Myocardial Thyroid Hormone Inactivating Deiodinase Results in Restrictive Cardiomyopathy in Mice. Mol. Endocrinol. 2012, 26, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Weltman, N.Y.; Ojamaa, K.; Schlenker, E.H.; Chen, Y.-F.; Zucchi, R.; Saba, A.; Colligiani, D.; Rajagopalan, V.; Pol, C.J.; Gerdes, A.M. Low-Dose T3 Replacement Restores Depressed Cardiac T3 Levels, Preserves Coronary Microvasculature and Attenuates Cardiac Dysfunction in Experimental Diabetes Mellitus. Mol. Med. 2014, 20, 302–312. [Google Scholar] [CrossRef]
- Russo, S.C.; Salas-Lucia, F.; Bianco, A.C. Deiodinases and the Metabolic Code for Thyroid Hormone Action. Endocrinology 2021, 162, 1–13. [Google Scholar] [CrossRef]
- Gereben, B.; Zavacki, A.M.; Ribich, S.; Kim, B.W.; Huang, S.A.; Simonides, W.S.; Zeöld, A.; Bianco, A.C. Cellular and Molecular Basis of Deiodinase-Regulated Thyroid Hormone Signaling. Endocr. Rev. 2008, 29, 898–938. [Google Scholar] [CrossRef] [Green Version]
- Janssen, R.; Zuidwijk, M.; Muller, A.; Mulders, J.; Oudejans, C.B.M.; Simonides, W.S. Cardiac Expression of Deiodinase Type 3 (Dio3) Following Myocardial Infarction Is Associated with the Induction of a Pluripotency MicroRNA Signature from the Dlk1-Dio3 Genomic Region. Endocrinology 2013, 154, 1973–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresham, K.S.; Mamidi, R.; Li, J.; Kwak, H.; Stelzer, J.E. Sarcomeric Protein Modification during Adrenergic Stress Enhances Cross-Bridge Kinetics and Cardiac Output. J. Appl. Physiol. 2017, 122, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, G.; Dolber, P.C.; Lefkowitz, R.J.; Koch, W.J. B-Adrenergic Receptor Kinase-1 Levels in Catecholamine-Induced Myocardial Hypertrophy. Hypertension 1999, 33, 396–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudej, R.K.; Iwase, M.; Uechi, M.; Vatner, D.E.; Oka, N.; Ishikawa, Y.; Shannon, R.P.; Bishop, S.P.; Vatner, S.F. Effects of Chronic β-Adrenergic Receptor Stimulation in Mice. J. Mol. Cell. Cardiol. 1997, 29, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Molojavyi, A.; Lindecke, A.; Raupach, A.; Moellendorf, S.; Köhrer, K.; Gödecke, A. Myoglobin-Deficient Mice Activate a Distinct Cardiac Gene Expression Program in Response to Isoproterenol-Induced Hypertrophy. Physiol. Genom. 2010, 41, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Saadane, N.; Alpert, L.; Chalifour, L.E. Expression of Immediate Early Genes, GATA-4, and Nkx-2.5 in Adrenergic-Induced Cardiac Hypertrophy and during Regression in Adult Mice. Br. J. Pharmacol. 1999, 127, 1165–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.J.C.; Rau, C.; Avetisyan, R.; Ren, S.; Romay, M.C.; Stolin, G.; Gong, K.W.; Wang, Y.; Lusis, A.J. Genetic Dissection of Cardiac Remodeling in an Isoproterenol-Induced Heart Failure Mouse Model. PLoS Genet. 2016, 12, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matkovich, S.J.; Diwan, A.; Klanke, J.L.; Hammer, D.J.; Marreez, Y.; Odley, A.M.; Brunskill, E.W.; Koch, W.J.; Schwartz, R.J.; Dorn, G.W. Cardiac-Specific Ablation of G-Protein Receptor Kinase 2 Redefines Its Roles in Heart Development and β-Adrenergic Signaling. Circ. Res. 2006, 99, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Lukowski, R.; Rybalkin, S.D.; Loga, F.; Leiss, V.; Beavo, J.A.; Hofmann, F. Cardiac Hypertrophy Is Not Amplified by Deletion of CGMP-Dependent Protein Kinase I in Cardiomyocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 5646–5651. [Google Scholar] [CrossRef] [Green Version]
- Drews, O.; Tsukamoto, O.; Liem, D.; Streicher, J.; Wang, Y.; Ping, P. Differential Regulation of Proteasome Function in Isoproterenol-Induced Cardiac Hypertrophy. Circ. Res. 2010, 107, 1094–1101. [Google Scholar] [CrossRef]
- Pol, C.J.; Muller, A.; Simonides, W.S. Cardiomyocyte-Specific Inactivation of Thyroid Hormone in Pathologic Ventricular Hypertrophy: An Adaptative Response or Part of the Problem. Heart Fail. Rev. 2010, 15, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gassanov, N.; Er, F.; Endres-Becker, J.; Wolny, M.; Schramm, C.; Hoppe, U.C. Distinct Regulation of Cardiac If Current via Thyroid Receptors Alpha1 and Beta1. Pflug. Arch. Eur. J. Physiol. 2009, 458, 1061–1068. [Google Scholar] [CrossRef]
- Schutkowski, A.; Wege, N.; Stangl, G.I.; König, B. Tissue-Specific Expression of Monocarboxylate Transporters during Fasting in Mice. PLoS ONE 2014, 9, e112118. [Google Scholar] [CrossRef] [Green Version]
- Escobar-Morreale, H.F.; Obregón, M.J.; Escobar del Rey, F.; Morreale de Escobar, G. Tissue-Specific Patterns of Changes in 3,5,3′-Triiodo-L-Thyronine Concentrations in Thyroidectomized Rats Infused with Increasing Doses of the Hormone. Which Are the Regulatory Mechanisms? Biochimie 1999, 81, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Thiele, R.H.; Nemergut, E.C.; Lynch, C. The Physiologic Implications of Isolated Alpha1 Adrenergic Stimulation. Anesth. Analg. 2011, 113, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boelen, A.; Wiersinga, W.M.; Fliers, E. Fasting-Induced Changes in the Hypothalamus-Pituitary-Thyroid Axis. Thyroid 2008, 18, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.A.; Stevenson, L.W.; Luu, M.; Walden, J.A. Altered Thyroid Hormone Metabolism in Advanced Heart Failure. J. Am. Coll. Cardiol. 1990, 16, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Ojamaa, K.; Kenessey, A.; Shenoy, R.; Klein, I. Thyroid Hormone Metabolism and Cardiac Gene Expression after Acute Myocardial Infarction in the Rat. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1319–E1324. [Google Scholar] [CrossRef]
- Henderson, K.K.; Danzi, S.; Paul, J.T.; Leya, G.; Klein, I.; Samarel, A.M. Physiological Replacement of T3 Improves Left Ventricular Function in an Animal Model of Myocardial Infarction-Induced Congestive Heart Failure. Circ. Heart Fail. 2009, 2, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Peeters, R.P.; Wouters, P.J.; Kaptein, E.; van Toor, H.; Visser, T.J.; Van den Berghe, G. Reduced Activation and Increased Inactivation of Thyroid Hormone in Tissues of Critically Ill Patients. J. Clin. Endocrinol. Metab. 2003, 88, 3202–3211. [Google Scholar] [CrossRef] [Green Version]
- Peeters, R.P.; Wouters, P.J.; van Toor, H.; Kaptein, E.; Visser, T.J.; Van den Berghe, G. Serum 3,3′,5′-Triiodothyronine (RT3) and 3,5,3′-Triiodothyronine/RT3 Are Prognostic Markers in Critically Ill Patients and Are Associated with Postmortem Tissue Deiodinase Activities. J. Clin. Endocrinol. Metab. 2005, 90, 4559–4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachucki, J.; Hopkins, J.; Peeters, R.; Tu, H.; Carvalho, S.D.; Kaulbach, H.; Abel, E.D.; Wondisford, F.E.; Ingwall, J.S.; Larsen, P.R. Type 2 Iodothyronin Deiodinase Transgene Expression in the Mouse Heart Causes Cardiac-Specific Thyrotoxicosis. Endocrinology 2001, 142, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Dentice, M.; Ambrosio, R.; Damiano, V.; Sibilio, A.; Luongo, C.; Guardiola, O.; Yennek, S.; Zordan, P.; Minchiotti, G.; Colao, A.; et al. Intracellular Inactivation of Thyroid Hormone Is a Survival Mechanism for Muscle Stem Cell Proliferation and Lineage Progression. Cell Metab. 2014, 20, 1038–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, M.J.; Banu, L.; Chen, Y.; Mandel, S.J.; Kieffer, J.D.; Harney, J.W.; Larsen, P.R. Recognition of UGA as a Selenocysteine Codon in Type I Deiodinase Requires Sequences in the 3′ Untranslated Region. Nature 1991, 353, 273–276. [Google Scholar] [CrossRef]
- Barefield, D.; Kumar, M.; de Tombe, P.P.; Sadayappan, S. Contractile Dysfunction in a Mouse Model Expressing a Heterozygous MYBPC3 Mutation Associated with Hypertrophic Cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2014, 306, 807–815. [Google Scholar] [CrossRef] [Green Version]
- Boelen, A.; van der Spek, A.H.; Bloise, F.; de Vries, E.M.; Surovtseva, O.V.; van Beeren, M.; Ackermans, M.T.; Kwakkel, J.; Fliers, E. Tissue Thyroid Hormone Metabolism Is Differentially Regulated during Illness in Mice. J. Endocrinol. 2017, 233, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Jongejan, R.M.S.; Klein, T.; Meima, M.E.; Visser, W.E.; van Heerebeek, R.E.A.; Luider, T.M.; Peeters, R.P.; de Rijke, Y.B. A Mass Spectrometry-Based Panel of Nine Thyroid Hormone Metabolites in Human Serum. Clin. Chem. 2020, 66, 556–566. [Google Scholar] [CrossRef]
- Kwakkel, J.; Chassande, O.; Van Beeren, H.C.; Fliers, E.; Wiersinga, W.M.; Boelen, A. Thyroid Hormone Receptor α Modulates Lipopolysaccharide-Induced Changes in Peripheral Thyroid Hormone Metabolism. Endocrinology 2010, 151, 1959–1969. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simonides, W.; Tijsma, A.; Boelen, A.; Jongejan, R.; de Rijke, Y.; Peeters, R.; Dentice, M.; Salvatore, D.; Muller, A. Divergent Thyroid Hormone Levels in Plasma and Left Ventricle of the Heart in Compensated and Decompensated Cardiac Hypertrophy Induced by Chronic Adrenergic Stimulation in Mice. Metabolites 2023, 13, 308. https://doi.org/10.3390/metabo13020308
Simonides W, Tijsma A, Boelen A, Jongejan R, de Rijke Y, Peeters R, Dentice M, Salvatore D, Muller A. Divergent Thyroid Hormone Levels in Plasma and Left Ventricle of the Heart in Compensated and Decompensated Cardiac Hypertrophy Induced by Chronic Adrenergic Stimulation in Mice. Metabolites. 2023; 13(2):308. https://doi.org/10.3390/metabo13020308
Chicago/Turabian StyleSimonides, Warner, Alice Tijsma, Anita Boelen, Rutchanna Jongejan, Yolanda de Rijke, Robin Peeters, Monica Dentice, Domenico Salvatore, and Alice Muller. 2023. "Divergent Thyroid Hormone Levels in Plasma and Left Ventricle of the Heart in Compensated and Decompensated Cardiac Hypertrophy Induced by Chronic Adrenergic Stimulation in Mice" Metabolites 13, no. 2: 308. https://doi.org/10.3390/metabo13020308