
Urinary Metabolic Distinction of Niemann–Pick Class 1 Disease through the Use of Subgroup Discovery

 , , ,
, , , 
Abstract
:1. Introduction
- Major Primary Objective: To employ SD technologies for the discovery of NPC1 disease subgroups with behaviours that are differentiable from those of the complete dataset;
- Secondary Objective (1): To provide urinary biomarker information, which is valuable for the diagnosis and prospective status monitoring of NPC1 disease;
- Secondary Objective (2): To explore these urinary biomarker subgroup discovery patterns in order to preliminarily detect any metabolic pathways that are impaired or disturbed in NPC1 disease (a process that may provide useful chemopathological and drug-targeting information).
2. Materials and Methods
2.1. Sample Collection, Preparation and Storage
2.2. 1H NMR Analysis of Urine Samples
2.3. Data Preprocessing
2.4. Urinary pH Values
2.5. Investigations of the Potential Influence of Added 2H2O on the pH Values of Urine Samples during Sample Preparation Stages
2.6. Analysing Data
2.6.1. Predictive Analysis
- The C4.5 algorithm [38] is the most known throughout the scientific literature and represents a decision-tree-generating algorithm that induces classification rules in this form from a set of given examples. C4.5 is based on the ID3 algorithm, and the main objective is to determine a decision tree that, on the basis of answers to questions regarding the input attributes, correctly predicts the identity or value of the target attribute.
- The FURIA algorithm [39] is a fuzzy rule learner based on the RIPPER implementation. FURIA does not use default rules, and it has special pruning procedures with respect to RIPPER. Its major objective is to extract a compact set of effective fuzzy rules from numerical data, and it has been shown to exhibit excellent behaviour in real-world problems with the same characteristics as the dataset analysed here.
- The k-NN algorithm [40] is the standard classification algorithm based on instances. The class of a given instance is assigned as the majority class with respect to its K closest instances according to a distance measure. The functioning of this algorithm is facile where, for example, for it to be classified, the K-nearest-neighbours method is applied. In this manner, the class proposed for the instance is the majority class in the very next vicinity of the instances where the vicinity is defined as the K instances with a lower distance for the instance to classify.
- The SMO [41] is a sequential minimal optimisation algorithm for training a support vector classifier, and its main objective is to build a support vector machine model with the training set, which then classifies all test data by means of the trained model using the SMO procedure.
2.6.2. Descriptive Analysis
- Interpretability. A SD proposal must obtain few rules containing a low number of variables in the antecedent part in order to help researchers to understand and use the extracted knowledge, i.e., simple and interpretable subgroups are preferred in the SD task.
- Trade-off sensitivity and confidence. These quality measures are relevant in SD because they indicate the percentage of positive examples covered, with the highest possible precision, respectively.
- Interest. Rules must provide unusual and interesting information within datasets. This objective is solved through the unusualness quality measure because it contributes to interest, generality and confidence in the problem.
2.7. Validation
2.8. Qualitative Over-Representation Network Enrichment Analysis (ORA)
3. Results
3.1. AUROC Results
3.2. Supervised Descriptive Rules Obtained by NMEEFSD
- For describing the rules obtained for this algorithm, it is important to highlight the following:
- Quality measures analysed for SD have a domain within the interval [0, 1], and these are relevant to measurements of the quality of the rules obtained with respect to trade-off between generality and precision, and interest. More information about these quality measures can be found in Appendix A.
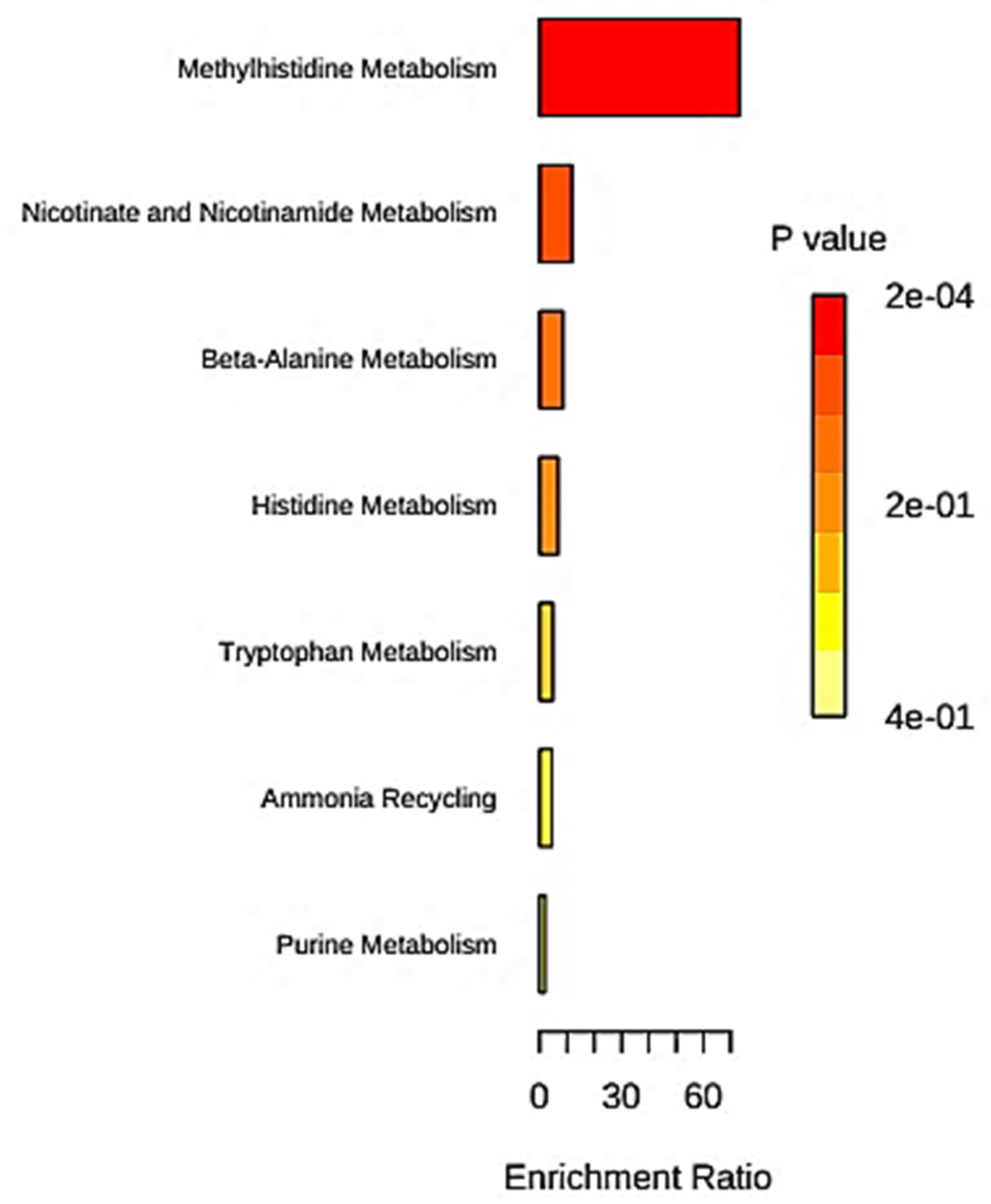
3.3. Over-Representation (Enrichment) Analysis
4. Discussion
4.1. Analysis from Subgroup Discovery
- All subgroups obtained have a low number of variables. For example, subgroups for the control heterozygous carrier class are between three and four variables, and for the NPC1 class, the subgroups have between six and seven variables. These values are low with respect to the whole dataset, which contains 54 continuous variables. This property shows the advantages of the use of this type of algorithm in order to analyse complex problems such as this one.
- The unusualness values are very interesting with values in the interval [0.55, 1.0]. As we have presented in ref. [50], we can indicate that all subgroups are contrasting and also serve as emerging rules. Specifically, it is interesting to note that values greater than 0.8 obtained for the subgroups of the NPC1 disease class show the unusualness and interest of the subgroups obtained.
- The relation between TPrate and fuzzy confidence is good. For the heterozygote class, this relation is excellent with values in confidence close to 1.0, along with excellent values in general. Nevertheless, in the NPC1 class, it should be noted that all or almost all examples of these collected samples are covered by the subgroups, respectively. However, despite the subgroups obtained in this class being specific, their confidence criterion values are somewhat lower.
- Finally, it should be considered that all values obtained for the TEF p value parameter are lower than the α = 0.10 considered in the experimental study, so all subgroups reject the null hypothesis, i.e., subgroups are interesting because there are significant differences between the proportions of positive and negative examples covered and not covered for each rule.
Potential Practical Applications of SGD, Including the Diagnosis and Prognosis of Diseases, and Therapeutic Interventions, Including Drug-Targeting Regimens
4.2. Metabolic Disturbances in NPC1 Disease Indicated by Imbalances in the Urinary 1H NMR Profiles of Patients with This Disorder
4.2.1. Tryptophan-Nicotinamide Metabolic Process
4.2.2. Kynurenine Pathway
4.2.3. Imbalances in Tryptophan Metabolism: Relevance to Lysosomal Storage Diseases
4.2.4. 3-Hydroxyphenylacetate and Tyrosine Metabolism
4.2.5. Significance of Further Non-Tryptophan-Nicotinamide/Tryptophan-Kynurenine Pathway Metabolites Featured in the SD Models Developed
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Quality Measures in Subgroup Discovery
- X = {Xm/m = 1, …, nv} is a set of features used to describe the subgroups, and nv is the number of descriptive features, e.g., a problem with nv = 3 such as Age, Sex and Visits.
- is the LL number of the variable nv, e.g., a representation of three linguistic labels for the Visits variable (V) where = Low, = Medium and = HighIn this manner, the quality measures analysed for this type of subgroups are:
- TPrate is the proportion of actual matches that have been classified correctly [83], and it has a component based on generality. It is computed as:
- FPrate is the proportion of instances that have been classified incorrectly for the nonclass of the rule. Its domain is [0, 1], and it is computed as:
- Fuzzy confidence is an adaptation of the standard confidence measure for fuzzy rules [84]. This quality measure obtains the precision of one subgroup. It has a domain [0, 1], and it is defined as:
Appendix B. Subgroup Discovery through Evolutionary Fuzzy Systems
References
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Walterfang, M.; Fahey, M.; Desmond, P.; Wood, A.; Seal, M.L.; Steward, C.; Adamson, C.; Kokkinos, C.; Fietz, M.; Velakoulis, D. White and gray matter alterations in adults with Niemann-Pick disease type C: A cross-sectional study. Neurology 2010, 75, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Wijburg, V.M.T.F. Recommendations for the diagnosis and management of Niemann–Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Garver, W.S.; Francis, G.A.; Jelinek, D.; Shepherd, G.; Flynn, J.; Castro, G.; Vockley, C.W.; Coppock, D.L.; Pettit, K.M.; Heidenreich, R.A.; et al. The national Niemann–Pick C1 disease database: Report of clinical features and health problems. Am. J. Med. Genet. Part A 2007, 143, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Percival, B.C.; Gibson, M.; Wilson, P.B.; Platt, F.M.; Grootveld, M. Metabolomic studies of lipid storage disorders, with special reference to Niemann-Pick type C disease: A critical review with future perspectives. Int. J. Mol. Sci. 2020, 21, 2533. [Google Scholar] [CrossRef]
- Kannenberg, F.; Nofer, J.R.; Schulte, E.; Reunert, J.; Marquardt, T.; Fobker, M. Determination of serum cholestane-3beta,5alpha,6beta-triol by gas chromatography-mass spectrometry for identification of Niemann-Pick type C (NPC) disease. J. Steroid Biochem. Mol. Biol. 2017, 169, 54–60. [Google Scholar] [CrossRef]
- Welford, R.W.; Garzotti, M.; Marques Lourenco, C.; Mengel, E.; Marquardt, T.; Reunert, J.; Amraoui, Y.; Kolb, S.A.; Morand, O.; Groenen, P. Plasma lysosphingomyelin demonstrates great potential as a diagnostic biomarker for Niemann-Pick disease type C in a retrospective study. PLoS ONE 2014, 9, e114669. [Google Scholar] [CrossRef]
- Jiang, X.; Sidhu, R.; Mydock-McGrane, L.; Hsu, F.F.; Covey, D.F.; Scherrer, D.E.; Earley, B.; Gale, S.E.; Farhat, N.Y.; Porter, F.D.; et al. Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci. Transl. Med. 2016, 8, 337ra63. [Google Scholar] [CrossRef]
- Sidhu, R.; Mondjinou, Y.; Qian, M.; Song, H.; Kumar, A.B.; Hong, X.; Hsu, F.F.; Dietzen, D.J.; Yanjanin, N.M.; Porter, F.D.; et al. N-acyl-O-phosphocholineserines: Structures of a novel class of lipids that are biomarkers for Niemann-Pick C1 disease. J. Lipid Res. 2019, 60, P1410–P1424. [Google Scholar] [CrossRef]
- Balboa, E.; Marín, T.; Oyarzún, J.E.; Contreras, P.S.; Hardt, R.; van den Bosch, T.; Alvarez, A.R.; Rebolledo-Jaramillo, B.; Klein, A.D.; Winter, D.; et al. Proteomic analysis of Niemann-Pick type C hepatocytes reveals potential therapeutic targets for liver damage. Cells 2021, 10, 2159. [Google Scholar] [CrossRef]
- Polo, G.; Burlina, A.; Furlan, F.; Kolamunnage, T.; Cananzi, M.; Giordano, L.; Zaninotto, M.; Plebani, M.; Burlina, A. High level of oxysterols in neonatal cholestasis: A pitfall in analysis of biochemical markers for Niemann-Pick type C disease. Clin. Chem. Lab. Med. 2016, 54, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.; Dunn, W.B.B.; Griffin, J.L.L.; Allwood, J.W.W.; Goodacre, R. Metabolic fingerprinting as a diagnostic tool. Pharmacogenetics 2007, 8, 1243–1266. [Google Scholar] [CrossRef] [PubMed]
- Wongravee, K.; Lloyd, G.R.; Silwood, C.J.; Grootveld, M.; Brereton, R.G. Supervised self organizing maps for classification and determination of potentially discriminatory variables: Illustrated by application to nuclear magnetic resonance metabolomic profiling. Anal. Chem. 2010, 82, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Lemanska, A.; Grootveld, M.; Silwood, C.J.; Brereton, R.G. Chemometric variance analysis of 1H NMR metabolomics data on the effects of oral rinse on saliva. Metabolomics 2011, 8, 64–80. [Google Scholar] [CrossRef]
- Camci, C.; Ersoy, C.; Kaynak, H. Abnormal respiratory event detection in sleep: A prescreening system with smart wearables. J. Biomed. Inform. 2019, 95, 103218. [Google Scholar] [CrossRef]
- Beunza, J.J.; Puertas, E.; Garcia-Ovejero, E.; Villalba, G.; Condes, E.; Koleva, G.; Hurtado, C.; Landecho, M.F. Comparison of machine learning algorithms for clinical event prediction (risk of coronary heart disease). J. Biomed. Inform. 2019, 97, 103257. [Google Scholar] [CrossRef]
- Pashazadeh, A.; Navimipour, N.J. Big data handling mechanisms in the healthcare applications: A comprehensive and systematic literature review. J. Biomed. Inform. 2018, 82, 47–62. [Google Scholar] [CrossRef]
- Herrera, F.; Carmona, C.J.; Gonzalez, P.; del Jesus, M.J. An overview on Subgroup Discovery: Foundations and Applications. Knowl. Inf. Syst. 2011, 29, 495–525. [Google Scholar] [CrossRef]
- Carmona, C.J.; Gonzalez, P.; del Jesus, M.J.; Herrera, F. Overview on evolutionary subgroup discovery: Analysis of the suitability and potential of the search performed by evolutionary algorithms. WIREs Data Min. Knowl. Discov. 2014, 4, 87–103. [Google Scholar] [CrossRef]
- Arrieta, A.B.; Díaz-Rodríguez, N.; Del Ser, J.; Bennetot, A.; Tabik, S.; Barbado, A.; Garcia, S.; Gil-Lopez, S.; Molina, D.; Benjamins, R.; et al. Explainable Artificial Intelligence (XAI): Concepts, taxonomies, opportunities and challenges toward responsible AI. Inf. Fusion 2020, 58, 82–115. [Google Scholar] [CrossRef]
- Carmona, C.J.; Chrysostomou, C.; Seker, H.; del Jesus, M.J. Fuzzy Rules for Describing Subgroups from Influenza A Virus Using a Multi-objective Evolutionary Algorithm. Appl. Soft Comput. 2013, 13, 3439–3448. [Google Scholar] [CrossRef]
- Carmona, C.J.; Gonzalez, P.; del Jesus, M.J.; Navıo, M.; Jimenez, L. Evolutionary Fuzzy Rule Extraction for Subgroup Discovery in a Psychiatric Emergency Department. Soft Comput. 2011, 15, 2435–2448. [Google Scholar] [CrossRef]
- Atzmueller, M.; Puppe, F.; Buscher, H.P. Towards Knowledge-Intensive Subgroup Discovery. In Proceedings of the Lernen—Wissensentdeckung—Adaptivitat—Fachgruppe Maschinelles Lernen, Hildesheim, Germany, 9–11 October 2004; pp. 111–117. [Google Scholar]
- Carmona, C.J.; Gonzalez, P.; del Jesus, M.J.; Herrera, F. NMEEF-SD: Non-dominated Multi-objective Evolutionary algorithm for Extracting Fuzzy rules in Subgroup Discovery. IEEE Trans. Fuzzy Syst. 2010, 18, 958–970. [Google Scholar] [CrossRef]
- Eiben, A.E.; Smith, J.E. Introduction to Evolutionary Computation; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Zadeh, L.A. The concept of a linguistic variable and its applications to approximate reasoning. Parts I, II, III. Inf. Sci. 1975, 8–9, 43–80, 199–249, 301–357. [Google Scholar] [CrossRef]
- Herrera, F. Genetic fuzzy systems: Taxomony, current research trends and prospects. Evol. Intell. 2008, 1, 27–46. [Google Scholar] [CrossRef]
- Fayyad, M.; Piatetsky-Shapiro, G.; Smyth, P. From data mining to knowledge discovery: An overview. In Advances in Knowledge Discovery and Data Mining; AAAI/MIT Press: Cambridge, MA, USA, 1996; pp. 1–34. [Google Scholar]
- Udert, K.M. The Fate of Nitrogen and Phosphorus in Source-Separated Urine. Ph.D. Thesis, Institute for Hydromechanics and Water Resources Management, Swiss Federal Institute of Technology, Zurich, Switzerland, 2003. [Google Scholar]
- Udert, K.M.; Larsen, T.A.; Gujer, W. Fate of major compounds in source-separated urine. Water Sci. Technol. 2004, 54, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Gloucestershire Hospitals NHS Foundation Trust. Citrate (Urine); Gloucestershire Hospitals NHS Foundation Trust: Gloucester, UK, 2019. [Google Scholar]
- Schreier, C.; Kremer, W.; Huber, F.; Neumann, S.; Pagel, P.; Lienemann, K.; Pestel, S. Reproducibility of NMR analysis of urine samples: Impact of sample preparation, storage conditions, and animal health status. BioMed Res. Int. 2013, 2013, 878374. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research (CDER), Food and Drug Administration, US Department of Health and Human Services; Center for Veterinary Medicine (CVM), Food and Drug Administration, US Department of Health and Human Services. Guidance for Industry. Bioanalytical Method Validation; US Department of Health and Human Services: Washington, DC, USA, 2001.
- Simerville, J.A.; Maxted, W.C.; Pahira, J.J. Urinalysis: A comprehensive review. Am. Fam. Physician 2005, 71, 1153–1162, Erratum in Am. Fam. Physician 2006, 74, 1096. [Google Scholar]
- Alcala’-Fdez, J.; Fernandez, A.; Luengo, J.; Derrac, J.; Garcıa, S.; Sanchez, L.; Herrera, F. KEEL Data-Mining Software Tool: Data Set Repository, Integration of Algorithms and Experimental Analysis Framework. J. Mult. Valued Log. Soft Comput. 2011, 17, 255–287. [Google Scholar]
- He, H.; Garcia, E. Learning from imbalanced data. IEEE Trans. Knowl. Data Eng. 2009, 21, 1263–1284. [Google Scholar]
- Sun, Y.; Wong, A.K.C.; Kamel, M. Classification of imbalanced data: A review. Int. J. Pattern Recognit. Artif. Intell. 2009, 23, 687–719. [Google Scholar] [CrossRef]
- Quinlan, J. C4.5: Programs for Machine Learning (Morgan Kaufmann Series in Machine Learning); Morgan Kaufmann: Burlington, MA, USA, 1993. [Google Scholar]
- Huhn, J.C.; Hullermeier, E. FURIA: An algorithm for unordered fuzzy rule induction. Data Min. Knowl. Discov. 2009, 19, 293–319. [Google Scholar] [CrossRef]
- McLachlan, G. Discriminant Analysis and Statistical Pattern Recognition; Wiley Series in Probability and Statistics; Wiley-Interscience: Hoboken, NJ, USA, 2004. [Google Scholar]
- Platt, J.C. Sequential Minimal Optimization: A Fast Algorithm for Training Support Vector Machines; MSR-TR-98-14; Microsoft: Redmond, DC, USA, 1998. [Google Scholar]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. Smote: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Bunkhumpornpat, C.; Sinapiromsaran, K.; Lursinsap, C. Safe-level-smote: Safe-level-synthetic minority over-sampling technique for handling the class imbalanced problem. In Proceedings of the 13th Pacific-Asia Conference on Advances in Knowledge Discovery and Data Mining, Bangkok, Thailand, 27–30 April 2009; pp. 475–482. [Google Scholar]
- Fisher, R.A. On the interpretation of χ2 from contingency tables, and the calculation of p. J. R. Stat. Soc. 1922, 85, 87–94. [Google Scholar] [CrossRef]
- Fisher, R.A. Statistical Methods for Research Workers, 20th ed.; Oliver and Boyd: Edinburgh, UK, 1954. [Google Scholar]
- Gamberger, D.; Lavrac, N. Expert-Guided Subgroup Discovery: Methodology and Application. J. Artif. Intell. Res. 2002, 17, 501–527. [Google Scholar] [CrossRef]
- Salzberg, S. A nearest hyperrectangle learning method. Mach. Learn. 1991, 6, 251–276. [Google Scholar] [CrossRef]
- Huang, J.; Ling, C. Using AUC and accuracy in evaluating learning algorithms. IEEE Trans. Knowl. Data Eng. 2005, 17, 299–310. [Google Scholar] [CrossRef]
- Hullermeier, E. Fuzzy sets in machine learning and data mining. Appl. Soft Comput. 2011, 11, 1493–1505. [Google Scholar] [CrossRef]
- Carmona, C.J.; del Jesus, M.J.; Herrera, F. A unifying analysis for the supervised descriptive rule discovery via the weighted relative accuracy. Knowl. Based Syst. 2018, 139, 89–100. [Google Scholar] [CrossRef]
- Kavsek, B.; Lavrac, N. APRIORI-SD: Adapting association rule learning to subgroup discovery. Appl. Artif. Intell. 2006, 20, 543–583. [Google Scholar] [CrossRef]
- Banerjee, D. Recent advances in the pathobiology of Hodgkin’s lymphoma: Potential impact on diagnostic, predictive, and therapeutic strategies. Adv. Hematol. 2011, 2011, 439456. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.O. Early-stage Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 363, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.E.; Yem, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Workman, P. Exploiting the cancer genome: Strategies for the discovery and clinical development of targeted molecular therapeutics. Ann. Rev. Pharmacol. Toxicol. 2012, 52, 549–573. [Google Scholar] [CrossRef]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef]
- Fukuwatari, T.; Shibata, K. Nutritional aspect of tryptophan metabolism. Int. J. Tryptophan Res. 2013, 6, 3–8. [Google Scholar] [CrossRef]
- Ruiz-Rodado, V.; Luque-Baena, R.M.; te Vruchte, D.; Probert, F.; Lachmann, R.H.; Hendriksz, C.J.; Wraith, J.E.; Imrie, J.; Elizondo, D.; Sillence, D.; et al. 1H NMR-linked urinary metabolic profiling of Niemann-Pick class C1 (NPC1) disease: Identification of potential new biomarkers using correlated component regression (CCR) and genetic algorithm (GA) analysis strategies. Curr. Metabolomics 2014, 2, 88–121. [Google Scholar] [CrossRef]
- Valle, M.; Price, R.; Nilsson, A.; Heyes, M.; Verotta, D. CSF quinolinic acid levels are determined by local HIV infection: Cross-sectional analysis and modeling of dynamics following antiretroviral therapy. Brain 2004, 127, 1047–1060. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequence. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy states. Int. J. Tryptophan Res. 2009, 2, S2097. [Google Scholar] [CrossRef]
- Widner, B.; Leblhuber, F.; Fuchs, D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J. Neural Transm. 2002, 109, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Sordillo, L.A.; Sordillos, P.P. Optical spectroscopy of tryptophan metabolites in neurodegenerative disease. In Neurophotonics and Biomedical Spectroscopy; Springer: Cham, Switzerland, 2019; pp. 137–157. [Google Scholar]
- Brautbar, A.; Elstein, D.; Pines, B.; Krienen, N.; Hemmer, J.; Buskila, D.; Zimran, A. Fibromyalgia and Gaucher’s disease. QJM Int. J. Med. 2005, 99, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Schwaz, M.J.; Offenbaecher, M.; Neumeister, A.; Ackenheil, M. Experimental evaluation of an altered tryptophan metabolism in fibromyalgia. Adv. Exp. Med. Biol. 2003, 527, 265–275. [Google Scholar]
- Harrison, B.J.; Olver, J.S.; Norman, T.R.; Nathan, P.J. Effects of serotonin and catecholamine depletion on interleukin-6 activation and mood in human volunteers. Hum. Psychopharmacol. 2002, 17, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Myer, B.J.; Khokher, A.M.; Rushton, N.; Cox, T.M. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: Increased release of interleukin-6 and interleukin-10. Q. J. Med. 1997, 90, 19–25. [Google Scholar] [CrossRef]
- Altarescu, G.; Phillips, M.; Foldes, A.J.; Elstein, D.; Zimran, A.; Mates, M. The interleukin-6 promoter polymorphism in Gaucher disease: A new modifier gene? Q. J. Med. 2003, 96, 575–578. [Google Scholar] [CrossRef]
- Davis, I.; Liu, A. What is the tryptophan kynurenine pathway and why is it important to neurotherapy? Expert Rev. Neurother. 2015, 15, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Roehrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Monch, E.; Kneer, J.; Jakobs, C.; Arnold, M.; Diehl, H.; Batzler, U. Examination of urine metabolites in the newborn period and during protein loading tests at 6 months of age (part 1). Eur. J. Pediatr. 1990, 149, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Rodado, V.; Nicoli, E.-R.; Probert, F.; Smith, D.A.; Morris, L.; Wassif, C.A.; Platt, F.M.; Grootveld, M. 1H NMR-linked metabolomics analysis of liver from a mouse model of NP-C1 disease. J. Proteome Res. 2016, 15, 3511–3527. [Google Scholar] [CrossRef]
- Konishi, Y. Transepithelial transport of microbial metabolites of quercetin in intestinal caco-2 cell monolayers. J. Agric. Food Chem. 2005, 53, 601–607. [Google Scholar] [CrossRef]
- Prior, R.L.; Rogers, T.R.; Khanal, R.C.; Wilkes, S.E.; Wu, X.; Howard, L.R. Urinary excretion of phenolic acids in rats fed cranberry. J. Agric. Food Chem. 2010, 58, 3940–3949. [Google Scholar] [CrossRef]
- Boudonck, K.J.; Mitchell, M.W.; Ne, M.L.S.; Keresztes, L.; Nyska, A.; Shinar, D.; Rosenstock, M. Discovery of metabolomics biomarkers for early detection of nephrotoxicity. Toxicol. Pathol. 2009, 37, 280–292. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 4, 608–617. [Google Scholar] [CrossRef]
- Song, D.-J.; Ho, Y.; Hsu, K.-Y. Metabolic kinetics of p-aminobenzoic acid in rabbits. Biopharm. Drug Dispos. 1999, 20, 263–270. [Google Scholar] [CrossRef]
- Lavrac, N.; Flach, P.A.; Zupan, B. Rule Evaluation Measures: A Unifying View. In Proceedings of the 9th International Workshop on Inductive Logic Programming, Bled, Slovenia, 24–27 June 1999; Springer: Berlin/Heidelberg, Germany, 1999; Volume 1634, pp. 174–185. [Google Scholar]
- Kloesgen, W. Explora: A Multipattern and Multistrategy Discovery Assistant. In Advances in Knowledge Discovery and Data Mining, American Association for Artificial Intelligence; American Association for Artificial Intelligence: Menlo Park, CA, USA, 1996; pp. 249–271. [Google Scholar]
- Del Jesus, M.J.; Gonzalez, P.; Herrera, F.; Mesonero, M. Evolutionary Fuzzy Rule Induction Process for Subgroup Discovery: A case study in marketing. IEEE Trans. Fuzzy Syst. 2007, 15, 578–592. [Google Scholar] [CrossRef]
- Doshi-Velez, F.; Kim, B. Towards a rigorous science of interpretable machine learning. arXiv 2017, arXiv:1702.08608. 2017. [Google Scholar]
- European Commission, Directorate-General for Communications Networks. Content and Technology, Ethics Guidelines for Trustworthy AI; Publications Office: Luxembourg, 2019; Available online: https://data.europa.eu/doi/10.2759/346720 (accessed on 10 June 2023).
- Holland, J.H. Adaptation in Natural and Artificial Systems; University of Michigan Press: Ann Arbor, MI, USA, 1975. [Google Scholar]
- Goldberg, D.E. Genetic Algorithms in Search, Optimization and Machine Learning; Addison-Wesley Longman Publishing Co., Inc.: Boston, MA, USA, 1989. [Google Scholar]
- Schwefel, H.P. Evolution and Optimum Seeking, Sixth-Generation Computer Technology Series; Wiley: Hoboken, NJ, USA, 1995. [Google Scholar]
- Fogel, D.B. Evolutionary Computation—Toward a New Philosophy of Machine Intelligence; IEEE Press: Piscataway, NJ, USA, 1995. [Google Scholar]
- Koza, J.R. Genetic Programming: On the Programming of Computers by Means of Natural Selection; MIT Press: Cambridge, MA, USA, 1992. [Google Scholar]
- Fernandez, A.; Garcıa, S.; Luengo, J.; Bernadó-Mansilla, E.; Herrera, F. Genetics-based machine learning for rule induction: State of the art, taxonomy, and comparative study. IEEE Trans. Evol. Comput. 2010, 14, 913–941. [Google Scholar] [CrossRef]
- Carmona, C.J.; Ruiz-Rodado, V.; del Jesus, M.J.; Weber, A.; Grootveld, M.; Gonzalez, P.; Elizondo, D. A fuzzy genetic programming-based algorithm for subgroup discovery and the application to one problem of pathogenesis of acute sore throat conditions in humans. Inf. Sci. 2015, 298, 180–197. [Google Scholar] [CrossRef]
- Carmona, C.J.; Ramırez-Gallego, S.; Torres, F.; Bernal, E.; del Jesus, M.J.; Garcıa, S. Web usage mining to improve the design of an e-commerce website: OrOliveSur.com. Expert Syst. Appl. 2012, 39, 11243–11249. [Google Scholar] [CrossRef]
- Carmona, C.J.; Gonzalez, P.; Garcıa-Domingo, B.; del Jesus, M.J.; Aguilera, J. MEFES: An evolutionary proposal for the detection of exceptions in subgroup discovery. An application to Concentrating Photovoltaic Technology. Knowl. Based Syst. 2013, 54, 73–85. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class/Prediction | Positive | Negative |
|---|---|---|
| Positive | True positive (TP) | False negative (FN) |
| Negative | False positive (FP) | True negative (TN) |
| Algorithm | Original | SLSMOTE |
|---|---|---|
| C4.5 | 0.6583 | 0.6833 |
| FURIA | 0.5791 | 0.5458 |
| k-NN | 0.5750 | 0.5958 |
| SMO | 0.5833 | 0.7041 |
| NMEEFSD | 0.5500 | 0.7125 |
| Rule | Description |
|---|---|
| R1 | IF Hippurate-C3/5-CH=normal AND Histidine-C2-CH=low AND Hypoxanthine-C3/5-CH=normal AND Quinolinate-C5-CH=normal THEN Heterozygote |
| R2 | IF Hippurate-C3/5-CH=normal AND Histidine-C2-CH=low AND Hypoxanthine-C3/5-CH=normal AND 1-Methylnicotinamide-C5-CH=low THEN Heterozygote |
| R3 | IF p-Aminobenzoate-C3/5-CH=low AND Hypoxanthine-C3/5-CH=normal AND Quinolinate-C5-CH=normal THEN Heterozygote |
| R4 | IF Hippurate-C3/5-CH=normal AND Hypoxanthine-C3/5-CH=normal AND 1-Methylnicotinamide-C5-CH=low THEN Heterozygote |
| R5 | IF Hippurate-C3/5-CH=normal AND p-Aminobenzoate-C3/5-CH=low AND Hypoxanthine-C3/5-CH=normal AND Quinolinate-C5-CH=normal THEN Heterozygote |
| R6 | IF Xanthurenate-C3-CH (s)=normal AND p-Aminobenzoate-C2/6-CH=normal AND p-Aminobenzoate-C3/5-CH=normal AND Hippurate-C2/6-CH=normal AND Quinaldate-C4-CH=normal AND Nicotinate-C2-CH=low AND Trigonelline-C2-CH=low THEN NPC1 |
| R7 | IF p-Aminobenzoate-C2/6-CH=normal AND Indoxylsulphate-C2/Phe-C2/6-CH=normal AND Hippurate-C2/6-CH=normal AND 3-Methylhistidine-C2-CH=normal AND Quinaldate-C4-CH=normal AND Trigonelline-C2-CH=low THEN NPC1 |
| Class | Rule | Vars | Unus | TPrate | FPrate | FCn f | TEF |
|---|---|---|---|---|---|---|---|
| R1 | 4 | 0.671 | 0.925 | 0.583 | 0.915 | 0.011 | |
| R2 | 4 | 0.796 | 0.675 | 0.083 | 0.967 | 0.000 | |
| Heterozygote | R3 | 3 | 0.708 | 0.750 | 0.333 | 0.971 | 0.014 |
| R4 | 3 | 0.767 | 0.700 | 0.167 | 0.960 | 0.001 | |
| R5 | 4 | 0.737 | 0.725 | 0.250 | 0.971 | 0.005 | |
| NPC1 | R6 | 7 | 0.800 | 1.000 | 0.400 | 0.403 | 0.000 |
| R7 | 6 | 0.808 | 0.917 | 0.300 | 0.484 | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona, C.J.; German-Morales, M.; Elizondo, D.; Ruiz-Rodado, V.; Grootveld, M. Urinary Metabolic Distinction of Niemann–Pick Class 1 Disease through the Use of Subgroup Discovery. Metabolites 2023, 13, 1079. https://doi.org/10.3390/metabo13101079
Carmona CJ, German-Morales M, Elizondo D, Ruiz-Rodado V, Grootveld M. Urinary Metabolic Distinction of Niemann–Pick Class 1 Disease through the Use of Subgroup Discovery. Metabolites. 2023; 13(10):1079. https://doi.org/10.3390/metabo13101079
Chicago/Turabian StyleCarmona, Cristóbal J., Manuel German-Morales, David Elizondo, Victor Ruiz-Rodado, and Martin Grootveld. 2023. "Urinary Metabolic Distinction of Niemann–Pick Class 1 Disease through the Use of Subgroup Discovery" Metabolites 13, no. 10: 1079. https://doi.org/10.3390/metabo13101079
APA StyleCarmona, C. J., German-Morales, M., Elizondo, D., Ruiz-Rodado, V., & Grootveld, M. (2023). Urinary Metabolic Distinction of Niemann–Pick Class 1 Disease through the Use of Subgroup Discovery. Metabolites, 13(10), 1079. https://doi.org/10.3390/metabo13101079