
Metabolic Alterations in NADSYN1-Deficient Cells

 ,
, _Verhoeven-Duif.png) and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of NADSYN1-Deficient HEK293T and A549 Cell Lines
2.2. Cell Lines
2.3. Intracellular NAD+ Depletion and Supplementation of (Labeled and Unlabeled) NAD+ Precursors
2.4. Metabolite Extraction for Metabolomics
2.5. LC-MS/MS Analysis of the Kynurenine Pathway
2.6. Untargeted Metabolomics by DI-HRMS
2.7. LC-MS/MS Analysis of the Glycolysis and Pentose Phosphate Pathway
2.8. GC-FID Analysis of Sugar Alcohols
2.9. Statistical Analysis
3. Results
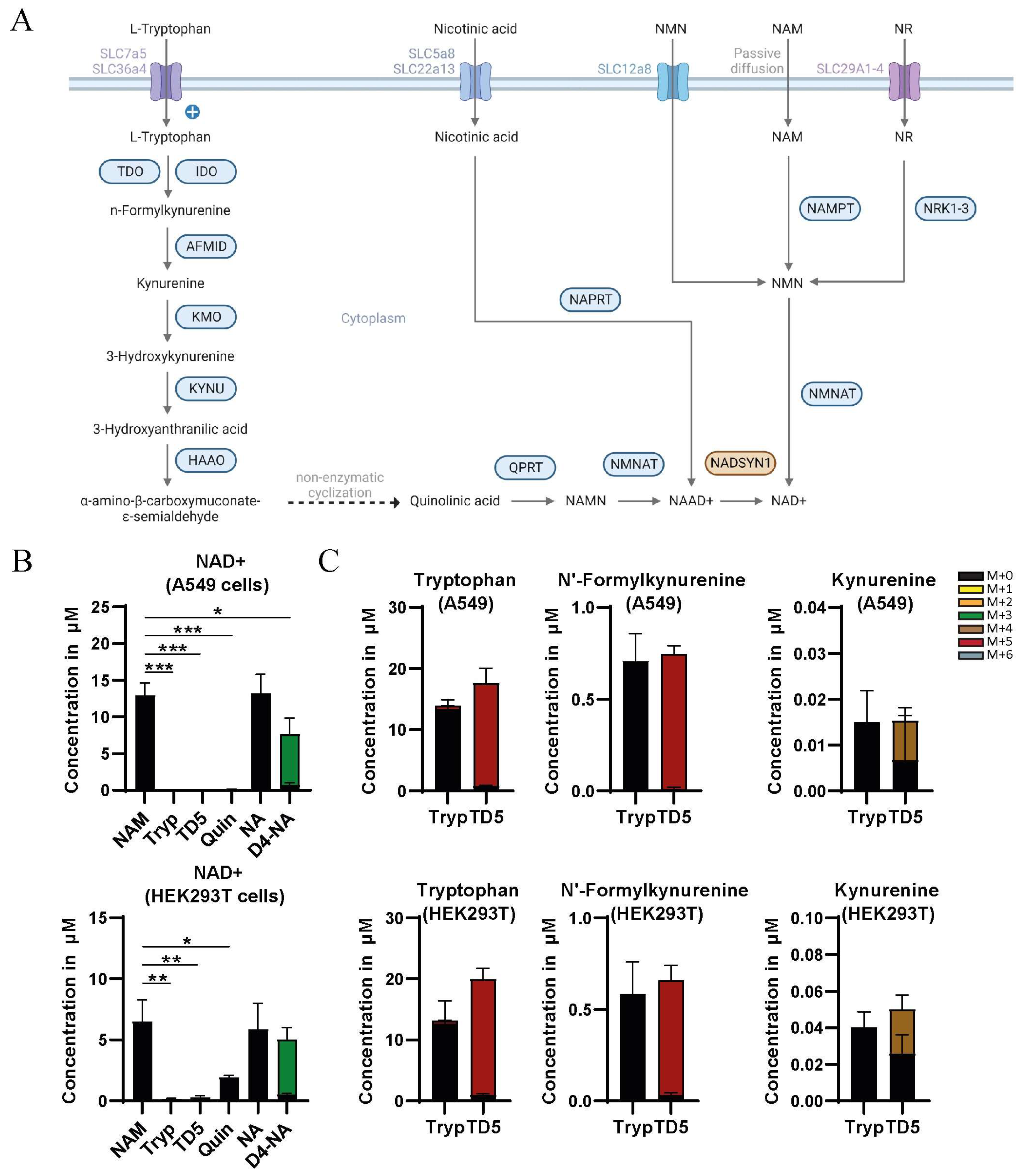
3.1. Nicotinamide and Nicotinic Acid but Not Tryptophan Are a Source of NAD+ in HEK293T and A549 Cells
3.2. Precursor Availability Determines Impact of NADSYN1 Deficiency
3.3. Reduced NAD+ Levels as a Consequence of NADSYN1 Deficiency Alter Flux through Glycolysis and Branching Pathways
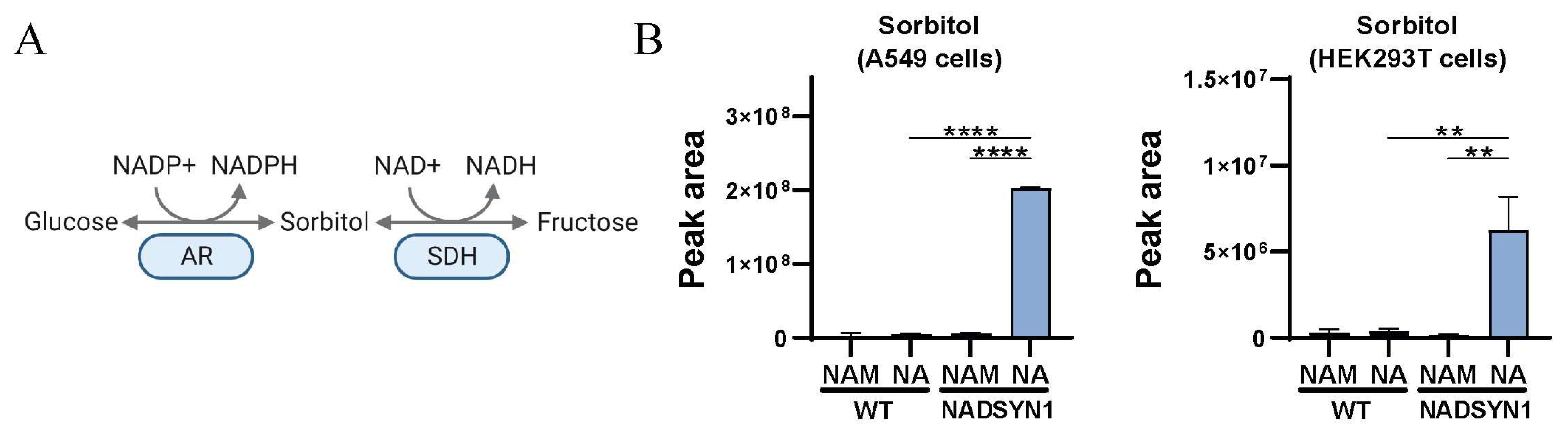
3.4. Reduced NAD+ Levels as a Consequence of NADSYN1 Deficiency Result in Accumulation of Sorbitol
4. Discussion
5. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef]
- Shi, H.; Enriquez, A.; Rapadas, M.; Martin, E.M.; Wang, R.; Moreau, J.; Lim, C.K.; Szot, J.O.; Ip, E.; Hughes, J.N.; et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N. Engl. J. Med. 2017, 377, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Szot, J.O.; Slavotinek, A.; Chong, K.; Brandau, O.; Nezarati, M.; Cueto-González, A.M.; Patel, M.S.; Devine, W.P.; Rego, S.; Acyinena, A.P.; et al. New cases that expand the genotypic and phenotypic spectrum of Congenital NAD Deficiency Disorder. Hum. Mutat. 2021, 42, 862–876. [Google Scholar] [CrossRef]
- Szot, J.O.; Campagnolo, C.; Cao, Y.; Iyer, K.R.; Cuny, H.; Drysdale, T.; Flores-Daboub, J.A.; Bi, W.; Westerfield, L.; Liu, P.; et al. Bi-allelic Mutations in NADSYN1 Cause Multiple Organ Defects and Expand the Genotypic Spectrum of Congenital NAD Deficiency Disorders. Am. J. Hum. Genet. 2020, 106, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhao, L.; Zhao, S.; Li, S.; Zhao, Z.; Chen, Z.; Zheng, Z.; Shao, J.; Niu, Y.; Li, X.; et al. Disruptive NADSYN1 Variants Implicated in Congenital Vertebral Malformations. Genes 2021, 12, 1615. [Google Scholar] [CrossRef] [PubMed]
- Kortbawi, H.; Ames, E.; Pritchard, A.; Devine, P.; van Ziffle, J.; Slavotinek, A. Further description of two patients with biallelic variants in NADSYN1 in association with cardiac and vertebral anomalies. Am. J. Med. Genet. A 2022, 188, 2479–2484. [Google Scholar] [CrossRef]
- Aubert-Mucca, M.; Janel, C.; Porquet-Bordes, V.; Patat, O.; Touraine, R.; Edouard, T.; Michot, C.; Tessier, A.; Cormier-Daire, V.; Attie-Bitach, T.; et al. Clinical heterogeneity of NADSYN1-associated VCRL syndrome. Clin. Genet. 2023, 104, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Erbs, E.; Brasen, C.L.; Lund, A.M.; Rasmussen, M. Adult patient diagnosed with NADSYN1 associated congenital NAD deficiency and analysis of NAD levels to be published in: European Journal of Medical Genetics. Eur. J. Med. Genet. 2023, 66, 104698. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Wang, H.; Majewski, J.; Wang, X.; Lopez, I.; Ren, H.; Chen, Y.; Li, Y.; Fishman, G.A.; Genead, M.; et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 2012, 44, 1035–1039. [Google Scholar] [CrossRef]
- Khan, A.O.; Budde, B.S.; Nürnberg, P.; Kawalia, A.; Lenzner, S.; Bolz, H.J. Genome-wide linkage and sequence analysis challenge CCDC66 as a human retinal dystrophy candidate gene and support a distinct NMNAT1-related fundus phenotype. Clin. Genet. 2018, 93, 149–154. [Google Scholar] [CrossRef]
- Nash, B.M.; Symes, R.; Goel, H.; Dinger, M.E.; Bennetts, B.; Grigg, J.R.; Jamieson, R.V. NMNAT1 variants cause cone and cone-rod dystrophy. Eur. J. Hum. Genet. 2018, 26, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Bedoni, N.; Quinodoz, M.; Pinelli, M.; Cappuccio, G.; Torella, A.; Nigro, V.; Testa, F.; Simonelli, F.; Corton, M.; Lualdi, S.; et al. An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs. Hum. Mol. Genet. 2020, 29, 2250–2260. [Google Scholar] [CrossRef] [PubMed]
- Bedoukian, E.C.; Zhu, X.; Serrano, L.W.B.; Scoles, D.; Aleman, T.S. Nmnat1-Associated Cone-Rod Dystrophy: Evidence for a Spectrum of Foveal Maldevelopment. Retin. Cases Brief Rep. 2022, 16, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, S.; Jayasena, T.; Richani, D.; Gilchrist, R.B.; Wu, L.E.; Sinclair, D.A.; Sachdev, P.S.; Braidy, N. Quantifying the cellular NAD+ metabolome using a tandem liquid chromatography mass spectrometry approach. Metabolomics 2017, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Cuny, H.; Kristianto, E.; Hodson, M.P.; Dunwoodie, S.L. Simultaneous quantification of 26 NAD-related metabolites in plasma, blood, and liver tissue using UHPLC-MS/MS. Anal. Biochem. 2021, 633, 114409. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Willemsen, M.; Van der Ham, M.; Gerrits, J.; Pras-Raves, M.L.; Prinsen, H.C.M.T.; Van Hasselt, P.M.; De Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites 2019, 9, 12. [Google Scholar] [CrossRef]
- Xiao, Y.; Elkins, K.; Durieux, J.K.; Lee, L.; Oeh, J.; Yang, L.X.; Liang, X.; DelNagro, C.; Tremayne, J.; Kwong, M.; et al. Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia 2013, 15, 1151–1160. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Shibata, T.; Osago, H.; Hashimoto, T.; Tsuchiya, M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 2007, 282, 24574–24582. [Google Scholar] [CrossRef]
- Cole, J.; Guiot, M.C.; Gravel, M.; Bernier, C.; Shore, G.C.; Roulston, A. Novel NAPRT specific antibody identifies small cell lung cancer and neuronal cancers as promising clinical indications for a NAMPT inhibitor/niacin co-administration strategy. Oncotarget 2017, 8, 77846–77859. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Pereira, S.; Pereira-Castro, I.; Silva, S.S.; Correia, M.G.; Neto, C.; da Costa, L.T.; Amorim, A.; Silva, R.M. Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors. Oncotarget 2016, 7, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Garavaglia, S.; Perozzi, S.; Galeazzi, L.; Raffaelli, N.; Rizzi, M. The crystal structure of human alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase in complex with 1,3-dihydroxyacetonephosphate suggests a regulatory link between NAD synthesis and glycolysis. FEBS J. 2009, 276, 6615–6623. [Google Scholar] [CrossRef] [PubMed]
- Huck, J.H.; Struys, E.A.; Verhoeven, N.M.; Jakobs, C.; van der Knaap, M.S. Profiling of pentose phosphate pathway intermediates in blood spots by tandem mass spectrometry: Application to transaldolase deficiency. Clin. Chem. 2003, 49, 1375–1380. [Google Scholar] [CrossRef]
- Eriksson, U.J.; Naeser, P.; Brolin, S.E. Increased accumulation of sorbitol in offspring of manifest diabetic rats. Diabetes 1986, 35, 1356–1363. [Google Scholar] [CrossRef]
- Eriksson, U.J.; Brolin, S.E.; Naeser, P. Influence of sorbitol accumulation on growth and development of embryos cultured in elevated levels of glucose and fructose. Diabetes Res. 1989, 11, 27–32. [Google Scholar]
- Cortese, A.; Zhu, Y.; Rebelo, A.P.; Negri, S.; Courel, S.; Abreu, L.; Bacon, C.J.; Bai, Y.; Bis-Brewer, D.M.; Bugiardini, E.; et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 2020, 52, 473–481. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meijer, N.W.F.; Gerrits, J.; Zwakenberg, S.; Zwartkruis, F.J.T.; Verhoeven-Duif, N.M.; Jans, J.J.M. Metabolic Alterations in NADSYN1-Deficient Cells. Metabolites 2023, 13, 1196. https://doi.org/10.3390/metabo13121196
Meijer NWF, Gerrits J, Zwakenberg S, Zwartkruis FJT, Verhoeven-Duif NM, Jans JJM. Metabolic Alterations in NADSYN1-Deficient Cells. Metabolites. 2023; 13(12):1196. https://doi.org/10.3390/metabo13121196
Chicago/Turabian StyleMeijer, Nils W. F., Johan Gerrits, Susan Zwakenberg, Fried J. T. Zwartkruis, Nanda M. Verhoeven-Duif, and Judith J. M. Jans. 2023. "Metabolic Alterations in NADSYN1-Deficient Cells" Metabolites 13, no. 12: 1196. https://doi.org/10.3390/metabo13121196
APA StyleMeijer, N. W. F., Gerrits, J., Zwakenberg, S., Zwartkruis, F. J. T., Verhoeven-Duif, N. M., & Jans, J. J. M. (2023). Metabolic Alterations in NADSYN1-Deficient Cells. Metabolites, 13(12), 1196. https://doi.org/10.3390/metabo13121196