
Chromosomal Instability Causes Sensitivity to Polyamines and One-Carbon Metabolism
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. RNA-Seq
2.2. Drosophila Stocks
2.3. Acridine Orange Staining
2.4. Immunostaining
2.5. Image Data Analysis and Statistics
2.6. Drug Treatments
2.7. ROS Detection and Quantification
3. Results
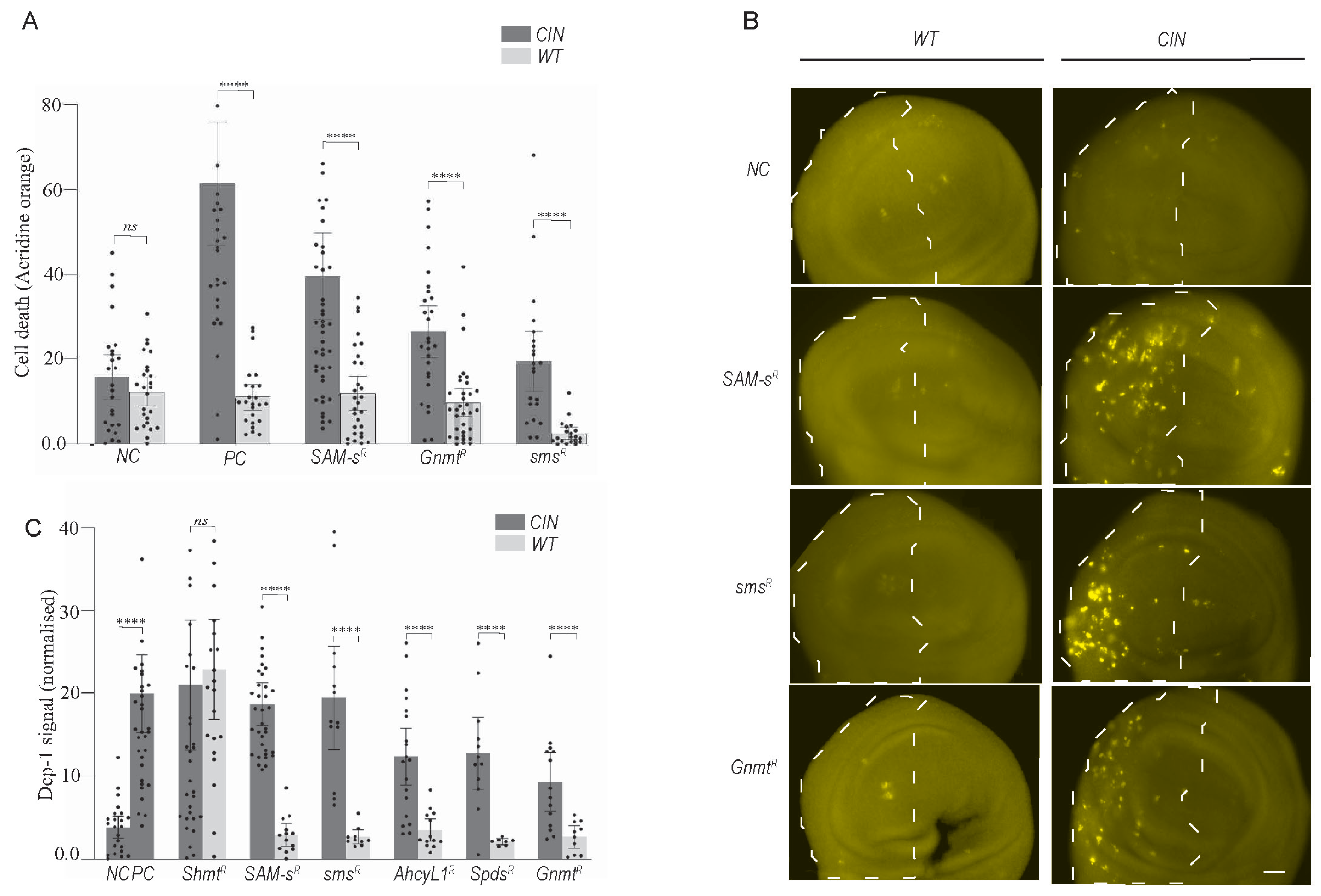
3.1. The Expression of One-Carbon Metabolism Genes Was Affected by Chromosomal Instability (CIN) and Affected CIN Tolerance
3.2. Knockdown of Candidates of One-Carbon and Polyamine Pathways Caused CIN Cell Death via Apoptosis
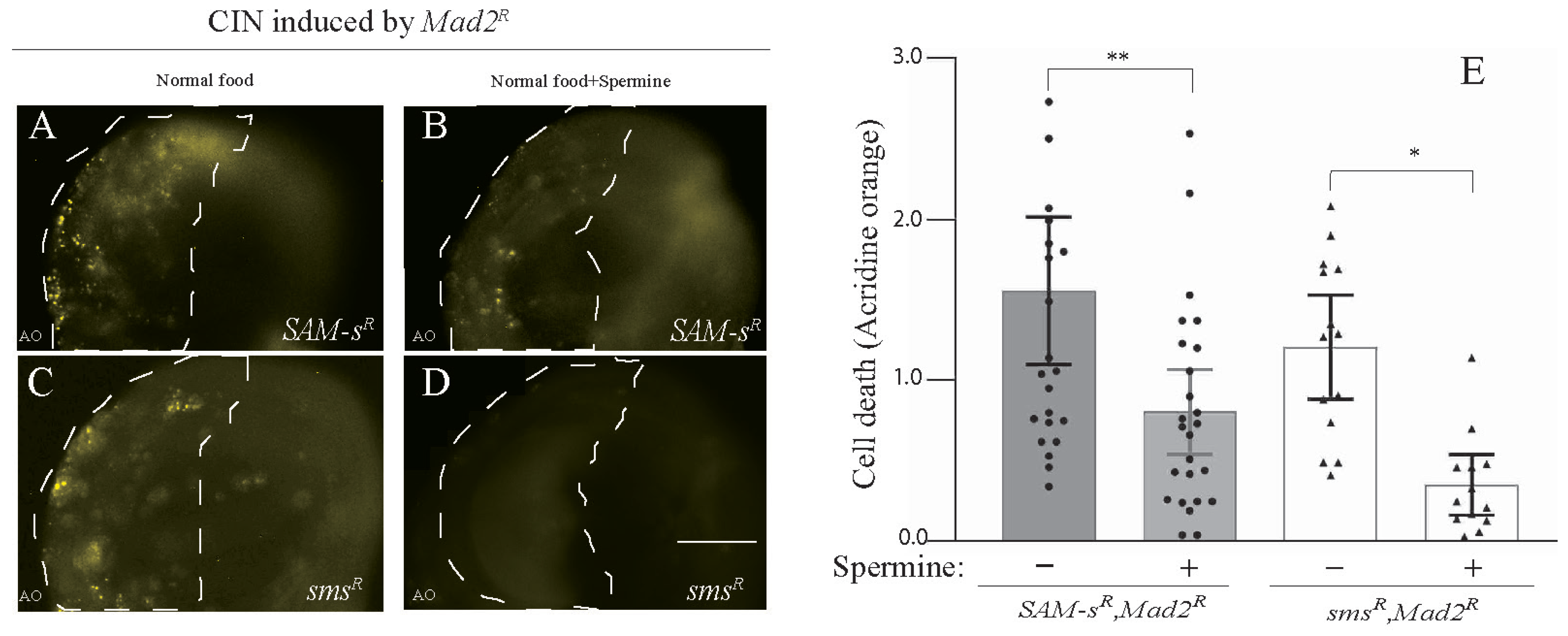
3.3. Feeding of Polyamines Rescued CIN Cell Death Caused by SAM-s or sms Depletion
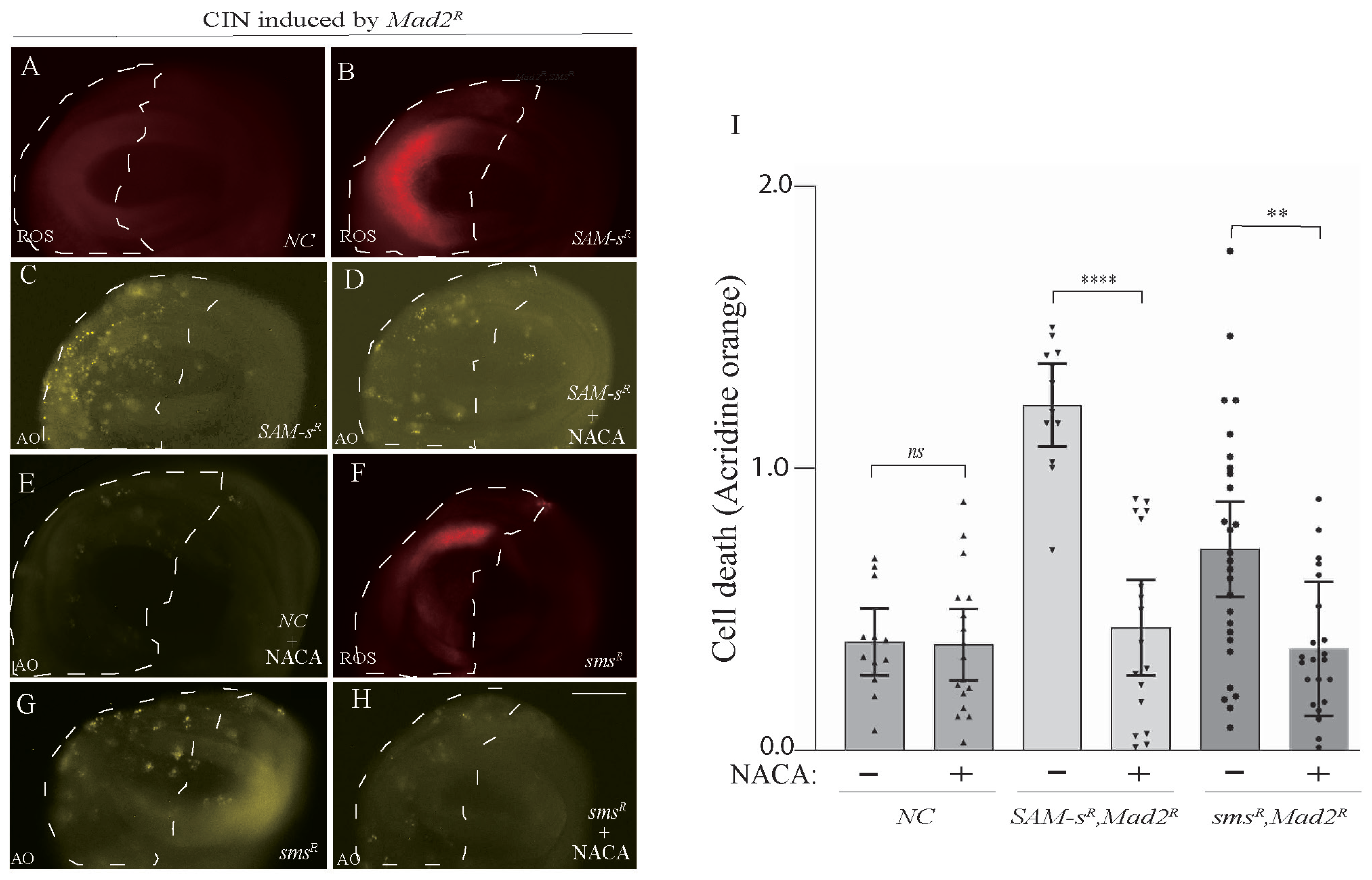
3.4. Antioxidant Feeding Rescued CIN Cell Death Phenotypes Caused by One-Carbon or Polyamine Metabolic Gene Depletion
3.5. CIN Cell Death from SAM-s and sms Knockdown Responded to Autophagy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of chromosomal instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Shaukat, Z.; Wong, H.W.; Nicolson, S.; Saint, R.B.; Gregory, S.L. A screen for selective killing of cells with chromosomal instability induced by a spindle checkpoint defect. PLoS ONE 2012, 7, e47447. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Shaukat, Z.; Saint, R.B.; Gregory, S.L. Chromosomal instability triggers cell death via local signalling through the innate immune receptor Toll. Oncotarget 2015, 6, 38552–38565. [Google Scholar] [CrossRef]
- Shaukat, Z.; Liu, D.; Choo, A.; Hussain, R.; O’Keefe, L.; Richards, R.; Saint, R.; Gregory, S.L. Chromosomal instability causes sensitivity to metabolic stress. Oncogene 2015, 34, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Shaukat, Z.; Xu, T.; Denton, D.; Saint, R.; Gregory, S. Autophagy regulates the survival of cells with chromosomal instability. Oncotarget 2016, 7, 63913–63923. [Google Scholar] [CrossRef]
- Kops, G.J.; Weaver, B.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef]
- Pfau, S.J.; Amon, A. Chromosomal instability and aneuploidy in cancer: From yeast to man. EMBO Rep. 2012, 13, 515–527. [Google Scholar] [CrossRef]
- Oromendia, A.B.; Dodgson, S.E.; Amon, A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012, 26, 2696–2708. [Google Scholar] [CrossRef]
- Cavuoto, P.; Fenech, M.F. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat Rev. 2012, 38, 726–736. [Google Scholar] [CrossRef]
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637. [Google Scholar] [CrossRef]
- Cavallaro, R.A.; Fuso, A.; Nicolia, V.; Scarpa, S. S-adenosylmethionine prevents oxidative stress and modulates glutathione metabolism in TgCRND8 mice fed a B-vitamin deficient diet. J. Alzheimers Dis. 2010, 20, 997–1002. [Google Scholar] [CrossRef]
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Shaukat, Z.; Saint, R.; Gregory, S.L. Chromosomal instability causes sensitivity to protein folding stress and ATP depletion. Biol. Open 2018, 7, bio038000. [Google Scholar] [CrossRef]
- Torres, E.M.; Dephoure, N.; Panneerselvam, A.; Tucker, C.M.; Whittaker, C.A.; Gygi, S.P.; Dunham, M.J.; Amon, A. Identification of aneuploidy-tolerating mutations. Cell 2010, 143, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Sheltzer, J.M.; Blank, H.M.; Pfau, S.J.; Tange, Y.; George, B.M.; Humpton, T.J.; Brito, I.L.; Hiraoka, Y.; Niwa, O.; Amon, A. Aneuploidy drives genomic instability in yeast. Science 2011, 333, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Dephoure, N.; Hwang, S.; O’Sullivan, C.; Dodgson, S.E.; Gygi, S.P.; Amon, A.; Torres, E.M. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife 2014, 3, e03023. [Google Scholar] [CrossRef]
- Wang, R.W.; Vigano, S.; Ben-David, U.; Amon, A.; Santaguida, S. Aneuploid senescent cells activate NF-kappaB to promote their immune clearance by NK cells. EMBO Rep. 2021, 22, e52032. [Google Scholar] [CrossRef]
- Kane, A.J.; Brennan, C.M.; Xu, A.E.; Solis, E.J.; Terhorst, A.; Denic, V.; Amon, A. Cell adaptation to aneuploidy by the environmental stress response dampens induction of the cytosolic unfolded-protein response. Mol. Biol. Cell 2021, 32, 1557–1564. [Google Scholar] [CrossRef]
- Obata, F.; Tsuda-Sakurai, K.; Yamazaki, T.; Nishio, R.; Nishimura, K.; Kimura, M.; Funakoshi, M.; Miura, M. Nutritional Control of Stem Cell Division through S-Adenosylmethionine in Drosophila Intestine. Dev. Cell 2018, 44, 741–751.e3. [Google Scholar] [CrossRef]
- Luka, Z.; Mudd, S.H.; Wagner, C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J. Biol. Chem. 2009, 284, 22507–22511. [Google Scholar] [CrossRef]
- Tain, L.S.; Jain, C.; Nespital, T.; Froehlich, J.; Hinze, Y.; Gronke, S.; Partridge, L. Longevity in response to lowered insulin signaling requires glycine N-methyltransferase-dependent spermidine production. Aging Cell 2020, 19, e13043. [Google Scholar] [CrossRef] [PubMed]
- Minarini, A.; Zini, M.; Milelli, A.; Tumiatti, V.; Marchetti, C.; Nicolini, B.; Falconi, M.; Farruggia, G.; Cappadone, C.; Stefanelli, C. Synthetic polyamines activating autophagy: Effects on cancer cell death. Eur. J. Med. Chem. 2013, 67, 359–366. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Vanrell, M.C.; Cueto, J.A.; Barclay, J.J.; Carrillo, C.; Colombo, M.I.; Gottlieb, R.A.; Romano, P.S. Polyamine depletion inhibits the autophagic response modulating Trypanosoma cruzi infectivity. Autophagy 2013, 9, 1080–1093. [Google Scholar] [CrossRef]
- Pietrocola, F.; Lachkar, S.; Enot, D.P.; Niso-Santano, M.; Bravo-San Pedro, J.M.; Sica, V.; Izzo, V.; Maiuri, M.C.; Madeo, F.; Marino, G.; et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015, 22, 509–516. [Google Scholar] [CrossRef]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef]
- Zhang, H.; Alsaleh, G.; Feltham, J.; Sun, Y.; Napolitano, G.; Riffelmacher, T.; Charles, P.; Frau, L.; Hublitz, P.; Yu, Z.; et al. Polyamines Control eIF5A Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol. Cell 2019, 76, 110–125 e119. [Google Scholar] [CrossRef] [PubMed]
- Warters, R.L.; Newton, G.L.; Olive, P.L.; Fahey, R.C. Radioprotection of human cell nuclear DNA by polyamines: Radiosensitivity of chromatin is influenced by tightly bound spermine. Radiat. Res. 1999, 151, 354–362. [Google Scholar] [CrossRef]
- Bale, S.; Ealick, S.E. Structural biology of S-adenosylmethionine decarboxylase. Amino Acids 2010, 38, 451–460. [Google Scholar] [CrossRef]
- Simile, M.M.; Saviozzi, M.; De Miglio, M.R.; Muroni, M.R.; Nufris, A.; Pascale, R.M.; Malvaldi, G.; Feo, F. Persistent chemopreventive effect of S-adenosyl-L-methionine on the development of liver putative preneoplastic lesions induced by thiobenzamide in diethylnitrosamine-initiated rats. Carcinogenesis 1996, 17, 1533–1537. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Nufris, A.; Daino, L.; Seddaiu, M.A.; Rao, P.M.; Rajalakshmi, S.; Sarma, D.S.; Feo, F. Chemoprevention by S-adenosyl-L-methionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis 1995, 16, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Garcea, R.; Pascale, R.; Daino, L.; Frassetto, S.; Cozzolino, P.; Ruggiu, M.E.; Vannini, M.G.; Gaspa, L.; Feo, F. Variations of ornithine decarboxylase activity and S-adenosyl-L-methionine and 5′-methylthioadenosine contents during the development of diethylnitrosamine-induced liver hyperplastic nodules and hepatocellular carcinoma. Carcinogenesis 1987, 8, 653–658. [Google Scholar] [CrossRef]
- Garcea, R.; Daino, L.; Pascale, R.; Simile, M.M.; Puddu, M.; Ruggiu, M.E.; Seddaiu, M.A.; Satta, G.; Sequenza, M.J.; Feo, F. Protooncogene methylation and expression in regenerating liver and preneoplastic liver nodules induced in the rat by diethylnitrosamine: Effect of variations of S-adenosylmethionine:S-adenosylhomocysteine ratio. Carcinogenesis 1989, 10, 1183–1192. [Google Scholar] [CrossRef]
- Garcea, R.; Daino, L.; Pascale, R.; Simile, M.M.; Puddu, M.; Frassetto, S.; Cozzolino, P.; Seddaiu, M.A.; Gaspa, L.; Feo, F. Inhibition of promotion and persistent nodule growth by S-adenosyl-L-methionine in rat liver carcinogenesis: Role of remodeling and apoptosis. Cancer Res. 1989, 49, 1850–1856. [Google Scholar] [PubMed]
- Feo, F.; Garcea, R.; Daino, L.; Pascale, R.; Pirisi, L.; Frassetto, S.; Ruggiu, M.E. Early stimulation of polyamine biosynthesis during promotion by phenobarbital of diethylnitrosamine-induced rat liver carcinogenesis. The effects of variations of the S-adenosyl-L-methionine cellular pool. Carcinogenesis 1985, 6, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Feo, F.; Garcea, R.; Pascale, R.; Pirisi, L.; Daino, L.; Donaera, A. The variations of S-adenosyl-L-methionine content modulate hepatocyte growth during phenobarbital promotion of diethylnitrosamine-induced rat liver carcinogenesis. Toxicol. Pathol. 1987, 15, 109–114. [Google Scholar] [CrossRef]
- Mahmood, N.; Cheishvili, D.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Pepin, A.S.; Szyf, M.; Rabbani, S.A. Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion, and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget 2018, 9, 5169–5183. [Google Scholar] [CrossRef] [PubMed]
- Shukeir, N.; Stefanska, B.; Parashar, S.; Chik, F.; Arakelian, A.; Szyf, M.; Rabbani, S.A. Pharmacological methyl group donors block skeletal metastasis in vitro and in vivo. Br. J. Pharmacol. 2015, 172, 2769–2781. [Google Scholar] [CrossRef]
- Parashar, S.; Cheishvili, D.; Arakelian, A.; Hussain, Z.; Tanvir, I.; Khan, H.A.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine blocks osteosarcoma cells proliferation and invasion in vitro and tumor metastasis in vivo: Therapeutic and diagnostic clinical applications. Cancer Med. 2015, 4, 732–744. [Google Scholar] [CrossRef]
- Kitada, M.; Xu, J.; Ogura, Y.; Monno, I.; Koya, D. Mechanism of Activation of Mechanistic Target of Rapamycin Complex 1 by Methionine. Front Cell Dev. Biol. 2020, 8, 715. [Google Scholar] [CrossRef]
- Dekanty, A.; Barrio, L.; Muzzopappa, M.; Auer, H.; Milan, M. Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc. Natl. Acad. Sci. USA 2012, 109, 20549–20554. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, A.; Shaukat, Z.; Newman, D.L.; Hussain, R.; Ricos, M.G.; Dibbens, L.; Gregory, S.L. Chromosomal Instability Causes Sensitivity to Polyamines and One-Carbon Metabolism. Metabolites 2023, 13, 642. https://doi.org/10.3390/metabo13050642
Islam A, Shaukat Z, Newman DL, Hussain R, Ricos MG, Dibbens L, Gregory SL. Chromosomal Instability Causes Sensitivity to Polyamines and One-Carbon Metabolism. Metabolites. 2023; 13(5):642. https://doi.org/10.3390/metabo13050642
Chicago/Turabian StyleIslam, Anowarul, Zeeshan Shaukat, David L. Newman, Rashid Hussain, Michael G. Ricos, Leanne Dibbens, and Stephen L. Gregory. 2023. "Chromosomal Instability Causes Sensitivity to Polyamines and One-Carbon Metabolism" Metabolites 13, no. 5: 642. https://doi.org/10.3390/metabo13050642