
The Over-Irradiation Metabolite Derivative, 24-Hydroxylumister-ol3, Reduces UV-Induced Damage in Skin
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Skin Explants
- Keratinocytes: Keratinocytes were grown out from skin fragments [75] and cultured as previously described with minor modifications [76], in keratinocyte growth medium (KGM), which contained minimum essential medium Eagle (M0518, Sigma-Aldrich, St. Louis, MO, USA), 0.02 M sodium bicarbonate (Sigma-Aldrich, St. Louis, MO, USA), 1 mM sodium pyruvate, and 25 mM HEPES (ThermoFisher Scientific, Waltham, MA, USA) in MilliQ water at pH 7.2 (Millipore SAS, Molsheim, France). Human primary keratinocytes were cultured in KGM containing 5% (v/v) fetal calf serum (FCS) and the following supplements: 5 μg/mL transferrin, 0.4 ug/mL hydrocortisone, 1 × 10−10 M cholera toxin, 10 ng/mL Epidermal Growth Factor (EGF), 5 μg/mL insulin (all from Sigma-Aldrich, St. Louis, MO, USA), and 2 × 10−11 M 3,3,5-triiodo-L-thyronine sodium salt [77]. Keratinocytes from passages 2–5 from at least two independent donors were used in all experiments. The keratinocyte culture media was changed to media without EGF and cholera toxin for 24 h before experiments to allow cells to become quiescent [78].
- Skin explants: Human skin explants were collected from consenting patients undergoing elective surgery at private hospitals in Sydney, Australia, and processed as previously described [56]. In brief, ice-cold sterile phosphate buffered saline (PBS) was used to transport the skin back to the laboratory. The skin was processed under aseptic conditions within 4 h of surgery. The skin was briefly washed with 4% chlorhexidine gluconate solution (Sigma-Aldrich, St. Louis, MO, USA) and then thoroughly washed with ice-cold PBS. Subcutaneous fat and debris were trimmed off to leave the epidermis and dermis only, for the study. The skin samples were dissected into 4 mm pieces with a punch biopsy tool with five biopsies prepared for each treatment. The skin samples were then prepared for UV irradiation as described below.
2.2. Solar-Simulated UV Irradiation
- Keratinocytes: Immediately prior to UV irradiation, the medium was replaced with irradiation buffer, Martinez solution containing 10 mM D-glucose (Sigma-Aldrich, St. Louis, MO, USA) without phenol red. For all keratinocyte experiments, the ssUV irradiation energy level was 400 mJ/cm2 UVB and 3600 mJ/cm2 UVA (4000 mJ/cm2) [53,79].
- Skin explants: Skin biopsies were placed in ice-cold sterile colorless Martinez buffer with the epidermis facing up in a volume that was just enough to surround the tissue without submersion, for UV radiation [56]. A single dose of ssUV at 20 J/cm2 was delivered to the skin explant samples. Sham/non-irradiated keratinocytes or skin were subjected to similar procedures but not irradiated.
2.3. 1,25( OH)2D3 and 24(OH)L3 Treatments
- Keratinocytes: Immediately after UV irradiation (or sham irradiation), the irradiation buffer was replaced with supplement-free keratinocyte growth medium (KGM) containing vehicle, 0.1% (v/v) ethanol, or treatments at the concentrations as indicated.
- Skin explants: Immediately after UV irradiation (or sham irradiation), the skin samples were treated with vehicle, 0.1% (v/v) ethanol, or treatments in RPMI-1640 media (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FCS, penicillin, and streptomycin at 37 °C.
2.4. Immunohistochemistry
- Keratinocytes:
- Skin explants:
2.5. Image Acquisition
- Keratinocytes: Bright-field images were acquired on the Olympus stereo investigator scope (MBF Bioscience, Williston, VT, USA) or on the Zeiss AxioScan-Z1 slide scanner (Zeiss Microscopy, Oberkochen, Germany) at the Bosch Advanced Microscopy Facility (University of Sydney). All images were taken at 20× magnification, and the immunohistochemical images were analyzed using ImageJ software. Using this program, images from an individual experiment were thresholded to the same value. The regions of interest (ROI) were randomly selected. The means and standard errors of the mean (SEMs) from three to five ROI per coverslip of each treatment were calculated and graphed. Staining and image analysis for CPDs and 8-OHdG produced similar results to those obtained using endonuclease detection of the lesion, followed by Comet assay [54,76].
- Skin explants: Bright-field images were captured using the Zeiss Axio Scan (Zeiss Microscopy, Oberkochen, Germany). For each section, images of the whole skin section were taken at 20× magnification. All images were analyzed using MetaMorph imaging software (Molecular Devices Corporation, San Jose, CA, USA), whereby the epidermal area was isolated and thresholded specific for positive nuclei in this region. This software then automatically calculated the positive nuclei as a percentage of the total epidermal area.
2.6. siRNA Transfection
2.7. Western Blot
2.8. Unscheduled DNA Synthesis
2.9. Seahorse Energetics
2.10. ATP Measurement
2.11. ROS Measurement
2.12. Statistical Analysis
- In vitro: Three independent experiments with triplicates per each treatment group were performed for each study with similar results, using keratinocytes from different donors. Unless otherwise indicated, the analyses were carried out by ANOVA with Tukey multiple comparisons post-test (GraphPad Prism statistical program) (CA, USA).
- Ex vivo: Three independent experiments were performed for each study with skin from different donors with similar results. Comparisons between treatments were made by one-way ANOVA followed by Sidak’s multiple comparisons test, using the GraphPad Prism statistical program (CA, USA), unless otherwise stated. The data in the graphs represent the mean + SEM, unless otherwise stated.
3. Results
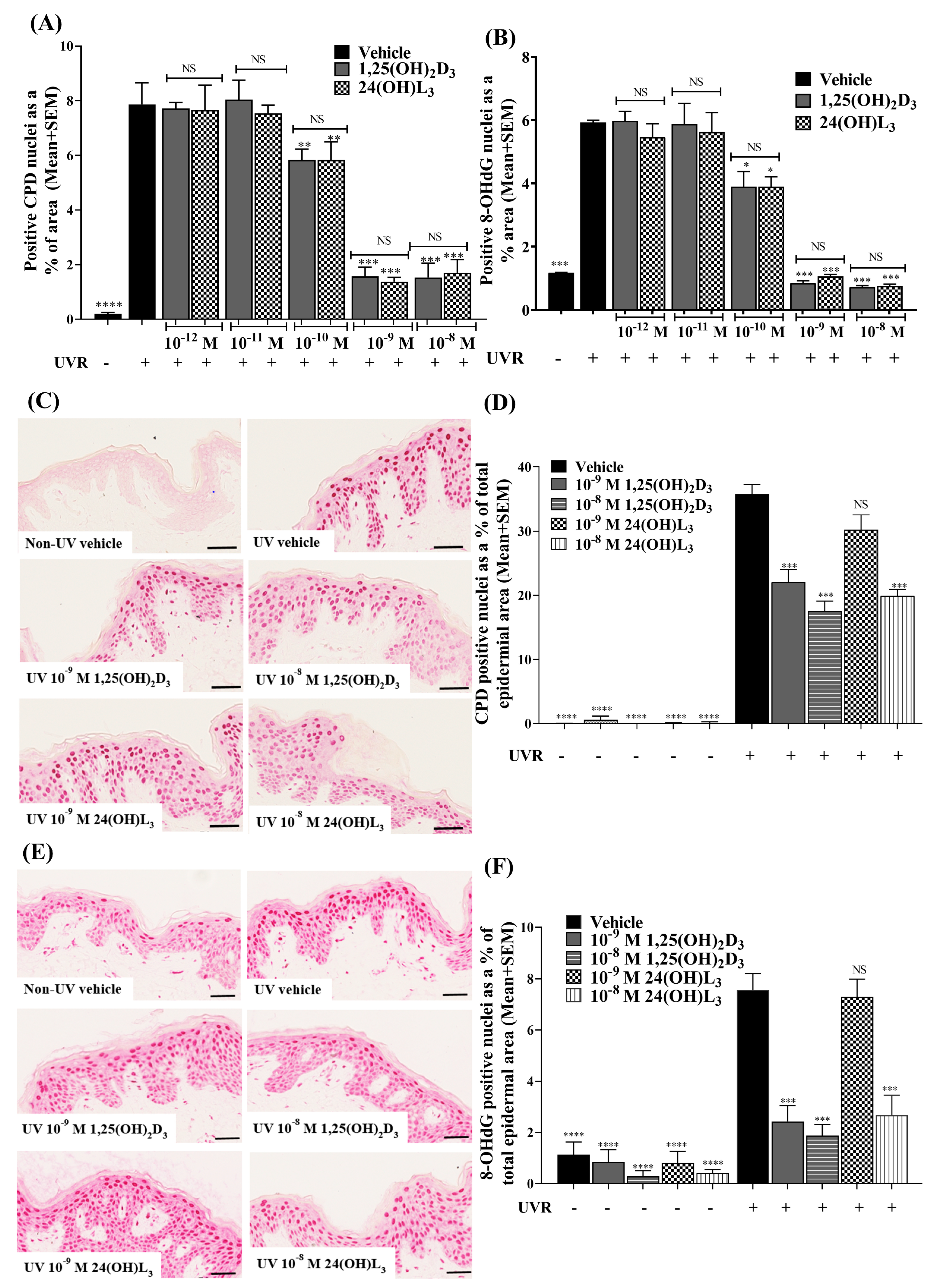
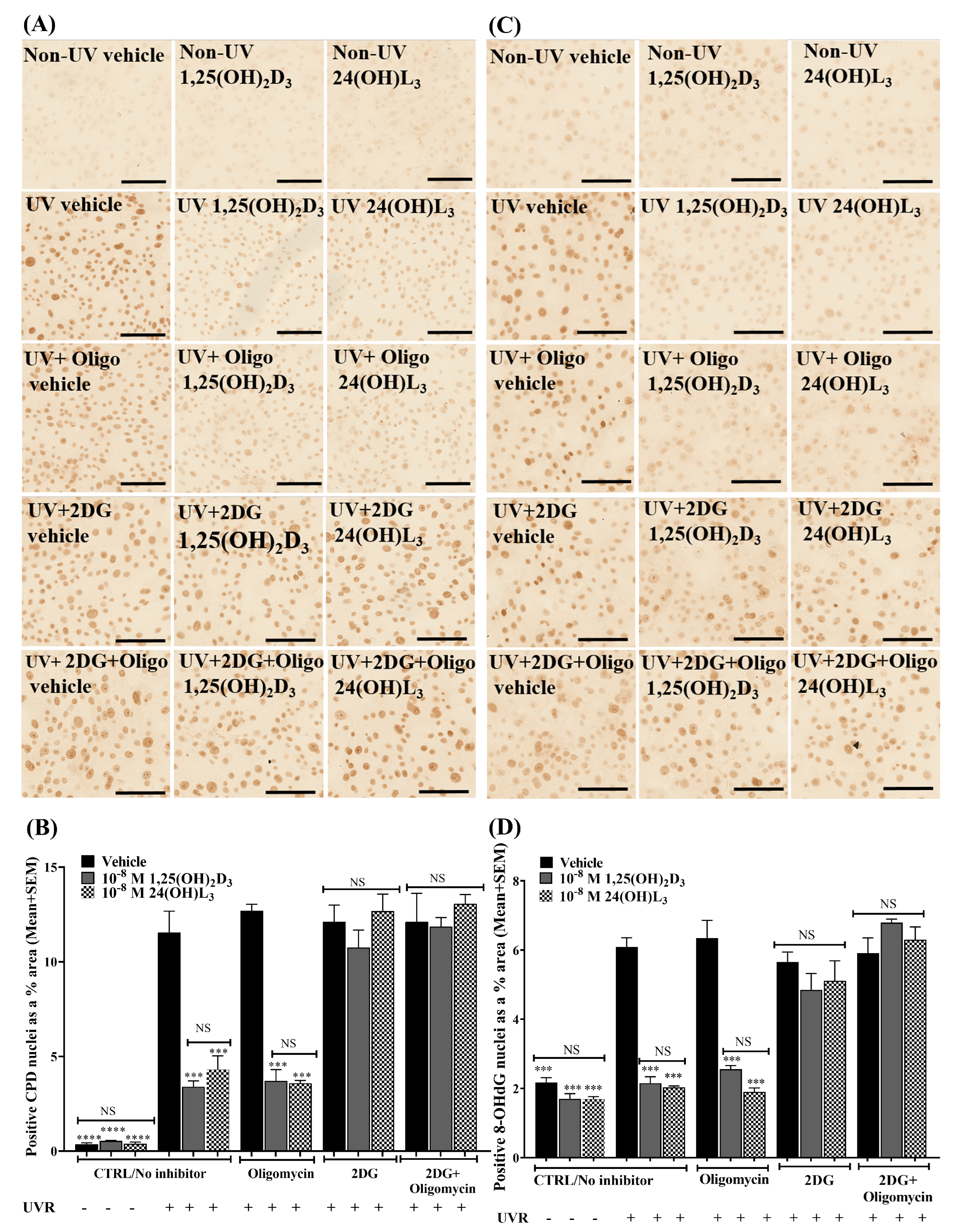
3.1. 24(OH)L3 Reduced UV-Induced CPDs and 8-OHdG in a Concentration-Dependent Manner, Similar to 1,25(OH)2D3 in Human Primary Keratinocytes
3.2. 24(OH)L3 Reduced UV-Induced CPDs and 8-OHdG at Higher Concentrations Than 1,25(OH)2D3 in Human Skin Explants
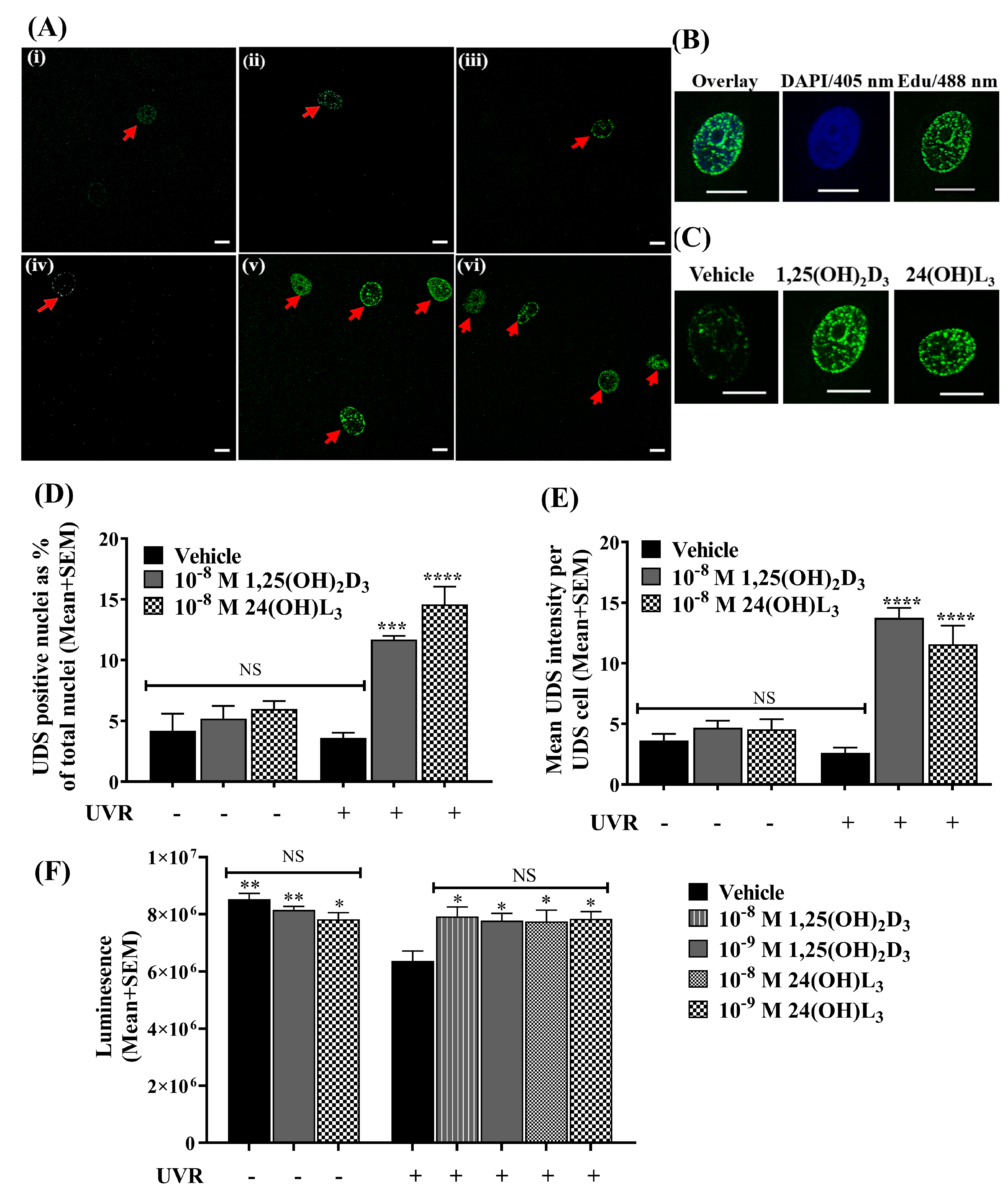
3.3. 24(OH)L3 Increased Unscheduled DNA Synthesis Similar to 1,25(OH)2D3 in UV-Irradiated Human Primary Keratinocytes
3.4. 24(OH)L3 Increased ATP Levels Similar to 1,25(OH)2D3 in UV-Irradiated Human Primary Keratinocytes
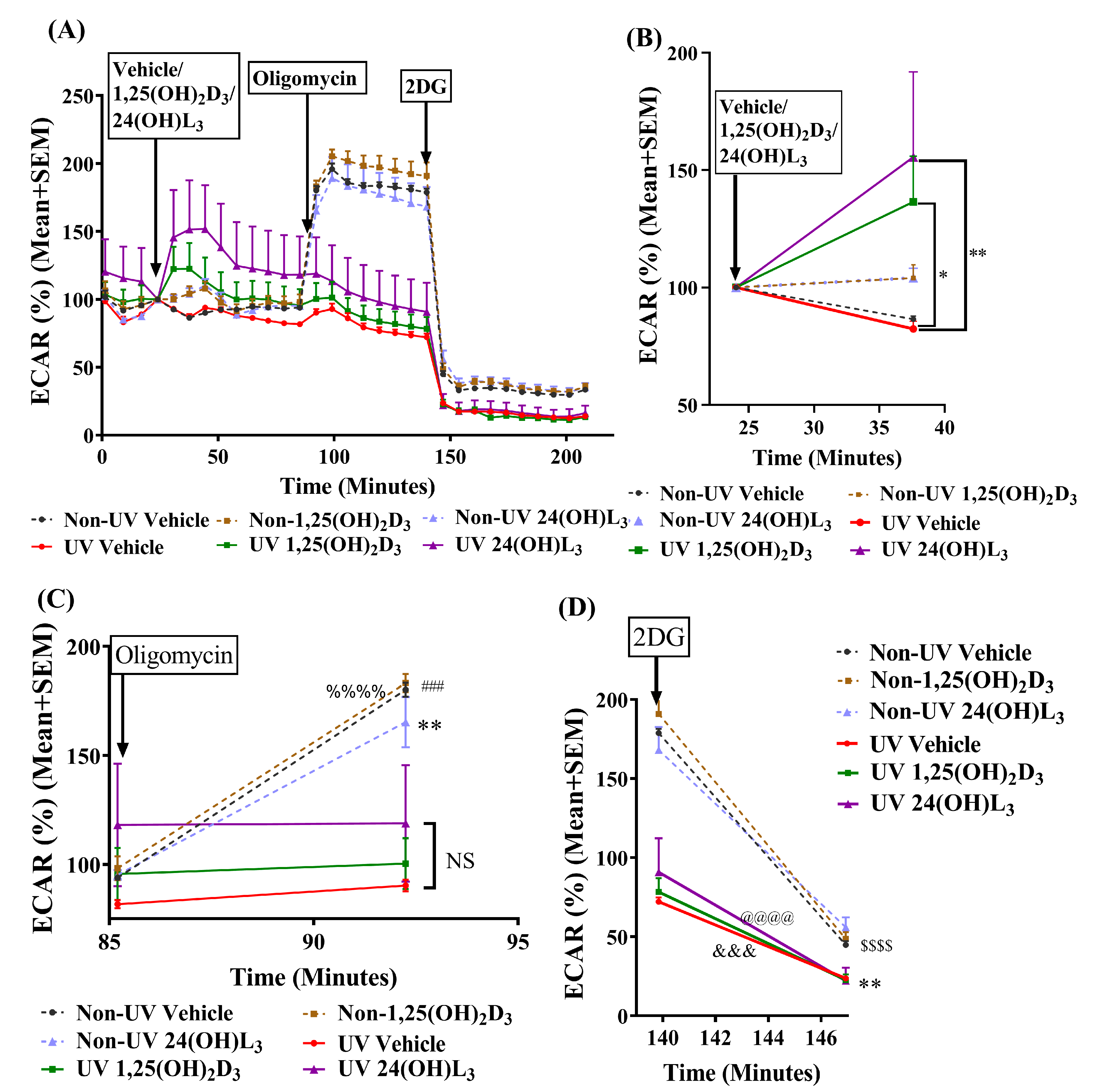
3.5. 24(OH)L3 Increased Glycolysis Similar to 1,25(OH)2D3 in UV-Irradiated Human Primary Keratinocytes
3.6. Reductions in UV-Induced CPDs and 8-OHdG by 24(OH)L3 or 1,25(OH)2D3 Were Abolished in the Presence of the Glycolysis Inhibitor (2-Deoxy-D-glucose) but Not by the Oxidative Phosphorylation Inhibitor (Oligomycin) in Human Primary Keratinocytes
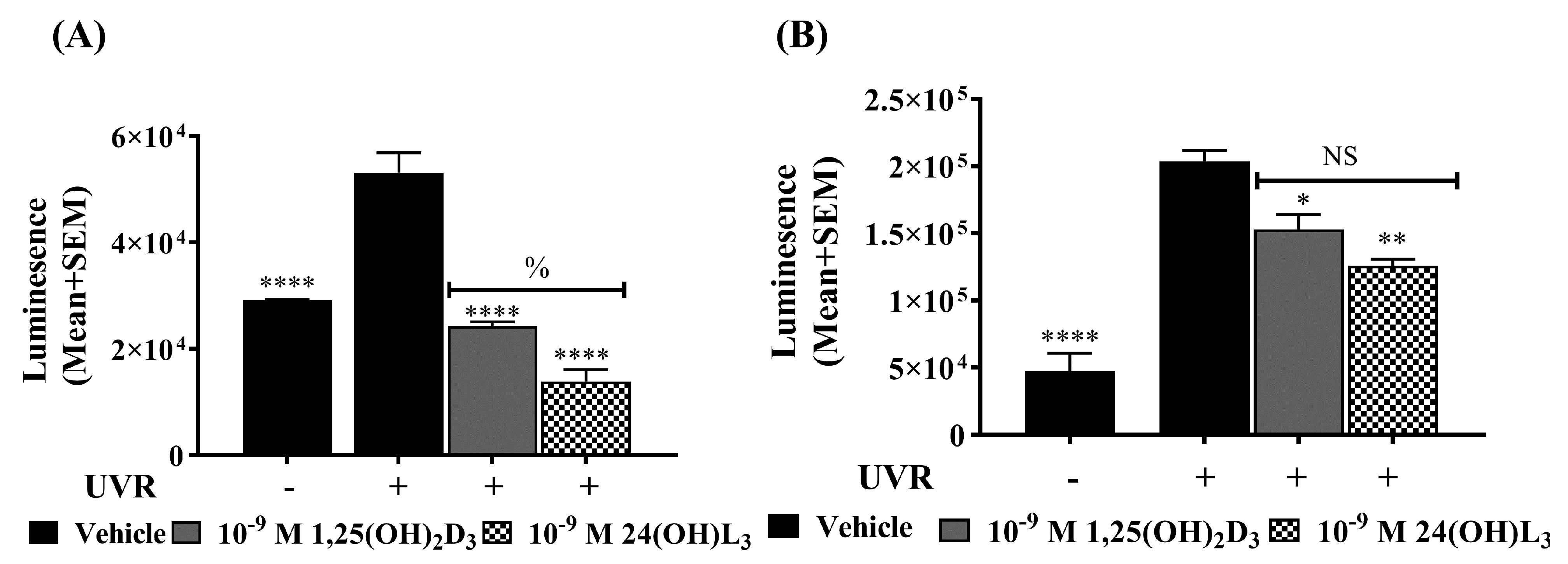
3.7. 24(OH)L3 Reduced Reactive Oxygen Species (ROS) Similar to 1,25(OH)2D3 in UV-Irradiated Human Primary Keratinocytes
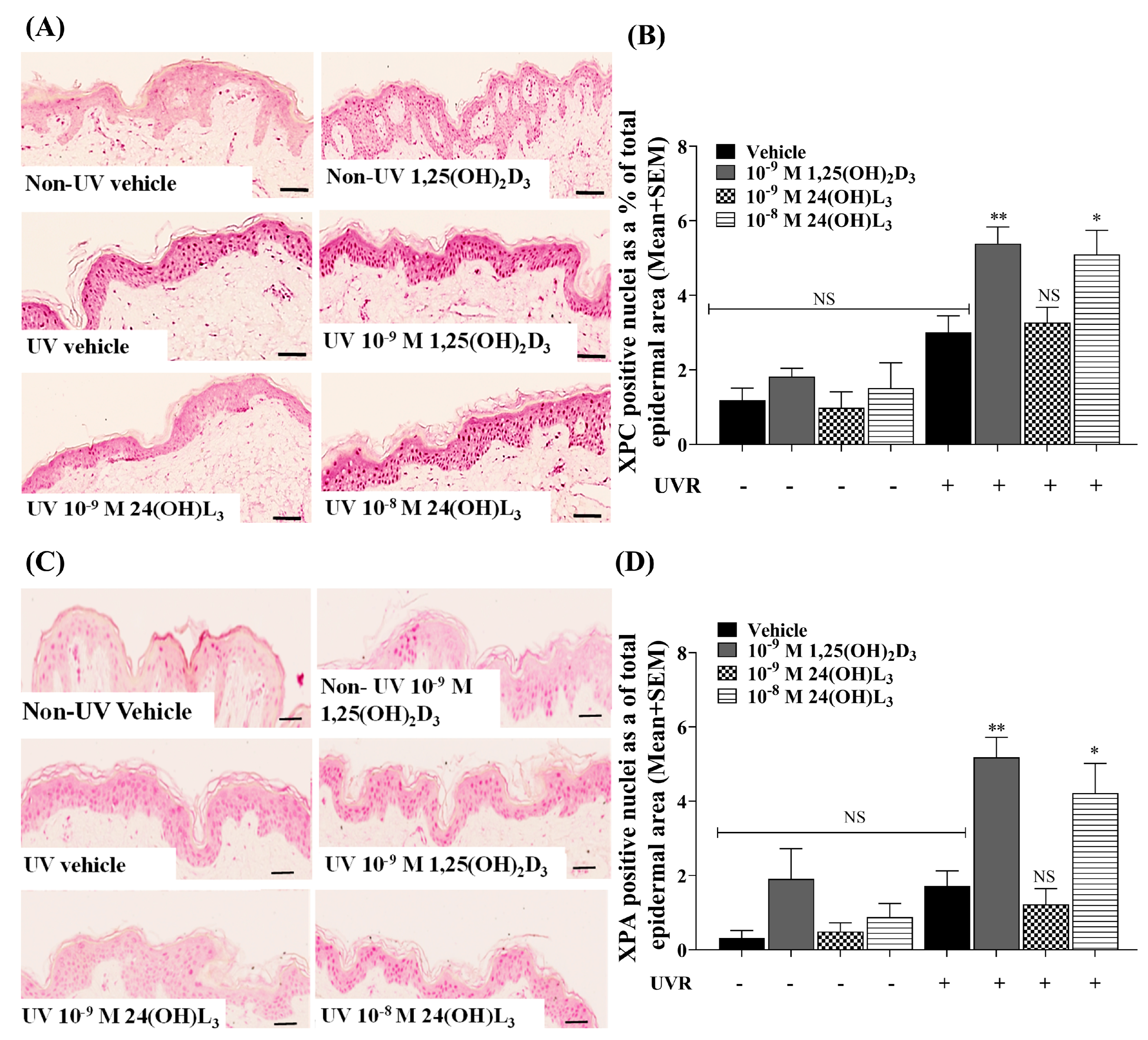
3.8. Topical Treatment with 1,25(OH)2D3 or 24(OH)L3 Increased XPC and XPA in UV-Irradiated Skin Explants
3.9. Reductions of UV-Induced CPDs by 1,25(OH)2D3 and 24(OH)L3 Were Abolished by Knockdown of XPC or XPA
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Urbach, F.; Forbes, P.D.; Davies, R.E.; Berger, D. Cutaneous Photobiology: Past, Present and Future. J. Investig. Dermatol. 1976, 67, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Beukers, R.; Berends, W. Isolation and identification of the irradiation product of thymine. Biochim. Biophys. Acta 1960, 41, 550–551. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Byrne, S.N.; Damian, D.L. Ultraviolet A radiation: Its role in immunosuppression and carcinogenesis. Semin. Cutan. Med. Surg. 2011, 30, 214–221. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ananthaswamy, H.N. Short-term and long-term cellular and molecular events following UV irradiation of skin: Implications for molecular medicine. Expert Rev. Mol. Med. 2004, 4, 1–22. [Google Scholar] [CrossRef]
- Amaro-Ortiz, A.; Yan, B.; D’Orazio, J.A. Ultraviolet radiation, aging and the skin: Prevention of damage by topical cAMP manipulation. Molecules 2014, 19, 6202–6219. [Google Scholar] [CrossRef]
- Yaar, M.; Gilchrest, B.A. Photoageing: Mechanism, prevention and therapy. Br. J. Dermatol. 2007, 157, 874–887. [Google Scholar] [CrossRef]
- Setlow, R.B.; Carrier, W.L. Identification of Ultraviolet-Induced Thymine Dimers in DNA by Absorbance Measurements. Photochem. Photobiol. 1963, 2, 49–57. [Google Scholar] [CrossRef]
- Douki, T.; Court, M.; Sauvaigo, S.; Odin, F.; Cadet, J. Formation of the Main UV-induced Thymine Dimeric Lesions within Isolated and Cellular DNA as Measured by High Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Biol. Chem. 2000, 275, 11678–11685. [Google Scholar] [CrossRef]
- Nakagawa, A.; Kobayashi, N.; Muramatsu, T.; Yamashina, Y.; Shirai, T.; Hashimoto, M.W.; Ikenaga, M.; Mori, T. Three-Dimensional Visualization of Ultraviolet-Induced DNA Damage and Its Repair in Human Cell Nuclei. J. Investig. Dermatol. 1998, 110, 143–148. [Google Scholar] [CrossRef]
- Ames, B.N.; Gold, L.S. Endogenous mutagens and the causes of aging and cancer. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1991, 250, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science 1983, 221, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Kvam, E.; Tyrrell, R.M. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis 1997, 18, 2379–2384. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Biological effects of the superoxide radical. Arch. Biochem. Biophys. 1986, 247, 1–11. [Google Scholar] [CrossRef]
- McAndrew, J.; Patel, R.P.; Jo, H.; Cornwell, T.; Lincoln, T.; Moellering, D.; White, C.R.; Matalon, S.; Darley-Usmar, V. The interplay of nitric oxide and peroxynitrite with signal transduction pathways: Implications for disease. Semin. Perinatol. 1997, 21, 351–366. [Google Scholar] [CrossRef]
- Leccia, M.T.; Yaar, M.; Allen, N.; Gleason, M.; Gilchrest, B.A. Solar simulated irradiation modulates gene expression and activity of antioxidant enzymes in cultured human dermal fibroblasts. Exp. Dermatol. 2001, 10, 272–279. [Google Scholar] [CrossRef]
- Podda, M.; Traber, M.G.; Weber, C.; Yan, L.-J.; Packer, L. UV-Irradiation Depletes Antioxidants and Causes Oxidative Damage in a Model of Human Skin. Free Radic. Biol. Med. 1998, 24, 55–65. [Google Scholar] [CrossRef]
- Van Loon, B.; Markkanen, E.; Hübscher, U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010, 9, 604–616. [Google Scholar] [CrossRef]
- Sander, C.S.; Chang, H.; Salzmann, S.; Müller, C.S.L.; Ekanayake-Mudiyanselage, S.; Elsner, P.; Thiele, J.J. Photoaging is Associated with Protein Oxidation in Human Skin In Vivo. J. Investig. Dermatol. 2002, 118, 618–625. [Google Scholar] [CrossRef]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Oxidative damage to DNA in mammalian chromatin. Mutat. Res. DNAging 1992, 275, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Agnez-Lima, L.F.; Melo, J.T.A.; Silva, A.E.; Oliveira, A.H.S.; Timoteo, A.R.S.; Lima-Bessa, K.M.; Martinez, G.R.; Medeiros, M.H.G.; Di Mascio, P.; Galhardo, R.S.; et al. DNA damage by singlet oxygen and cellular protective mechanisms. Mutat. Res. Rev. Mutat. Res. 2012, 751, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Treoule, R. Comparative study of oxidation of nucleic acid components by hydroxyl radicals, singlet oxygen and superoxide anion radicals. Photochem. Photobiol. 1978, 28, 661–667. [Google Scholar] [CrossRef]
- Hallett, F.R.; Hallett, B.P.; Snipes, W. Reactions between Singlet Oxygen and the Constituents of Nucleic Acids: Importance of Reactions in Photodynamic Processes. Biophys. J. 1970, 10, 305–315. [Google Scholar] [CrossRef]
- Kasai, H.; Crain, P.F.; Kuchino, Y.; Nishimura, S.; Ootsuyama, A.; Tanooka, H. Formation of 8-hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis 1986, 7, 1849–1851. [Google Scholar] [CrossRef]
- Kasai, H.; Nishimura, S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984, 12, 2137–2145. [Google Scholar] [CrossRef]
- Hei, T.K.; Liu, S.X.; Waldren, C. Mutagenicity of arsenic in mammalian cells: Role of reactive oxygen species. Proc. Natl. Acad. Sci. USA 1998, 95, 8103–8107. [Google Scholar] [CrossRef]
- Lynn, S.; Gurr, J.-R.; Lai, H.-T.; Jan, K.-Y. NADH Oxidase Activation Is Involved in Arsenite-Induced Oxidative DNA Damage in Human Vascular Smooth Muscle Cells. Circ. Res. 2000, 86, 514–519. [Google Scholar] [CrossRef]
- Simm, A.; Brömme, H.-J. Reactive oxygen species (ROS) and aging: Do we need them—Can we measure them—Should we block them? Signal Transduct. 2005, 5, 115–125. [Google Scholar] [CrossRef]
- Birch-Machin, M.A.; Swalwell, H. How mitochondria record the effects of UV exposure and oxidative stress using human skin as a model tissue. Mutagenesis 2010, 25, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Bosch, R.; Philips, N.; Suárez-Pérez, J.; Juarranz, A.; Devmurari, A.; Chalensouk-Khaosaat, J.; González, S. Mechanisms of Photoaging and Cutaneous Photocarcinogenesis, and Photoprotective Strategies with Phytochemicals. Antioxidants 2015, 4, 248. [Google Scholar] [CrossRef] [PubMed]
- Ogura, R.; Sugiyama, M.; Nishi, J.; Haramaki, N. Mechanism of Lipid Radical Formation Following Exposure of Epidermal Homogenate to Ultraviolet Light. J. Investig. Dermatol. 1991, 97, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Bau, D.T.; Gurr, J.R.; Jan, K.Y. Nitric oxide is involved in arsenite inhibition of pyrimidine dimer excision. Carcinogenesis 2001, 22, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Feig, D.I.; Reid, T.M.; Loeb, L.A. Reactive Oxygen Species in Tumorigenesis. Cancer Res. 1994, 54, 1890s–1894s. [Google Scholar] [PubMed]
- De Laat, W.L.; Jaspers, N.G.J.; Hoeijmakers, J.H.J. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785. [Google Scholar] [CrossRef]
- Hansson, J.; Munn, M.; Rupp, W.D.; Kahn, R.; Wood, R.D. Localization of DNA repair synthesis by human cell extracts to a short region at the site of a lesion. J. Biol. Chem. 1989, 264, 21788–21792. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Zotter, A.; Luijsterburg, M.S.; Warmerdam, D.O.; Ibrahim, S.; Nigg, A.; Van Cappellen, W.A.; Hoeijmakers, J.H.J.; Van Driel, R.; Vermeulen, W.; Houtsmuller, A.B. Recruitment of the Nucleotide Excision Repair Endonuclease XPG to Sites of UV-Induced DNA Damage Depends on Functional TFIIH. Mol. Cell. Biol. 2006, 26, 8868–8879. [Google Scholar] [CrossRef]
- Costa, R.M.A.; Chiganças, V.; Da Silva Galhardo, R.; Carvalho, H.; Menck, C.F.M. The eukaryotic nucleotide excision repair pathway. Biochimie 2003, 85, 1083–1099. [Google Scholar] [CrossRef]
- Aboussekhra, A.; Biggerstaff, M.; Shivji, M.K.K.; Vilpo, J.A.; Moncollin, V.; Podust, V.N.; Protić, M.; Hübscher, U.; Egly, J.-M.; Wood, R.D. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 1995, 80, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Okamoto, T.; Shimizu, Y.; Masutani, C.; Iwai, S.; Hanaoka, F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001, 15, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Ng, J.M.Y.; Masutani, C.; Iwai, S.; Van der Spek, P.J.; Eker, A.P.M.; Hanaoka, F.; Bootsma, D.; Hoeijmakers, J.H.J. Xeroderma Pigmentosum Group C Protein Complex Is the Initiator of Global Genome Nucleotide Excision Repair. Mol. Cell 1998, 2, 223–232. [Google Scholar] [CrossRef]
- Van Cuijk, L.; Vermeulen, W.; Marteijn, J.A. Ubiquitin at work: The ubiquitous regulation of the damage recognition step of NER. Exp. Cell Res. 2014, 329, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Naegeli, H.; Sugasawa, K. The xeroderma pigmentosum pathway: Decision tree analysis of DNA quality. DNA Repair 2011, 10, 673–683. [Google Scholar] [CrossRef]
- Ikegami, T.; Kuraoka, I.; Saijo, M.; Kodo, N.; Kyogoku, Y.; Morikawa, K.; Tanaka, K.; Shirakawa, M. Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nat. Struct. Mol. Biol. 1998, 5, 701. [Google Scholar] [CrossRef]
- Fadda, E. Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Comput. Struct. Biotechnol. J. 2016, 14, 78–85. [Google Scholar] [CrossRef]
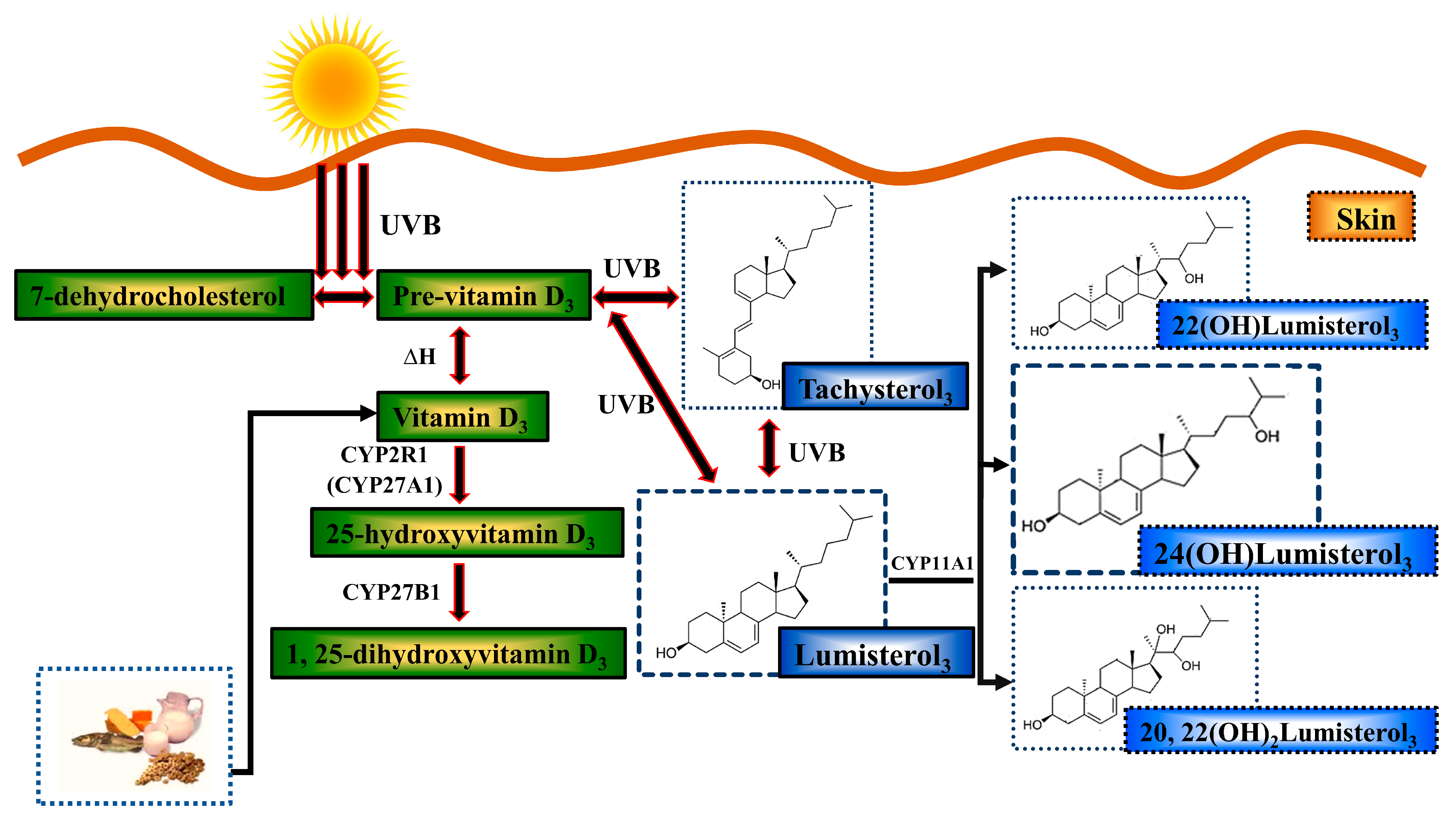
- Holick, M.F. The Cutaneous Photosynthesis of Previtamin D3: A Unique Photoendocrine System. J. Investig. Dermatol. 1981, 77, 51–58. [Google Scholar] [CrossRef]
- Bikle, D.D.; Nemanic, M.K.; Gee, E.; Elias, P. 1,25-Dihydroxyvitamin D3 production by human keratinocytes. Kinetics and regulation. J. Clin. Investig. 1986, 78, 557–566. [Google Scholar] [CrossRef]
- Bikle, D.D.; Nemanic, M.K.; Whitney, J.O.; Elias, P.W. Neonatal human foreskin keratinocytes produce 1,25-dihydroxyvitamin D3. Biochemistry 1986, 25, 1545–1548. [Google Scholar] [CrossRef]
- Lehmann, B.; Sauter, W.; Knuschke, P.; Dreßler, S.; Meurer, M. Demonstration of UVB-induced synthesis of 1α,25-dihydroxyvitamin D3 (calcitriol) in human skin by microdialysis. Arch. Dermatol. Res. 2003, 295, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.M.; Deo, S.S.; Wong, G.; Slater, M.; Norman, A.W.; Bishop, J.E.; Posner, G.H.; Ishizuka, S.; Halliday, G.M.; Reeve, V.E.; et al. Skin cancer prevention: A possible role of 1,25dihydroxyvitamin D3 and its analogs. J. Steroid Biochem. Mol. Biol. 2005, 97, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.M.; Norman, A.W.; Sequeira, V.B.; Mohan, R.; Rybchyn, M.S.; Reeve, V.E.; Halliday, G.M.; Mason, R.S. 1α,25(OH)2-Vitamin D and a Nongenomic Vitamin D Analogue Inhibit Ultraviolet Radiation–Induced Skin Carcinogenesis. Cancer Prev. Res. 2011, 4, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Thomson, C.; Gupta, R.; Tongkao-on, W.; Ryan, A.; Halliday, G.M.; Mason, R.S. 1[small alpha],25 Dihydroxyvitamin D3 enhances cellular defences against UV-induced oxidative and other forms of DNA damage in skin. Photochem. Photobiol. Sci. 2012, 11, 1837–1847. [Google Scholar] [CrossRef]
- Gupta, R.; Dixon, K.M.; Deo, S.S.; Holliday, C.J.; Slater, M.; Halliday, G.M.; Reeve, V.E.; Mason, R.S. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J. Investig. Derm. 2007, 127, 707–715. [Google Scholar] [CrossRef]
- Song, E.J.; Gordon-Thomson, C.; Cole, L.; Stern, H.; Halliday, G.M.; Damian, D.L.; Reeve, V.E.; Mason, R.S. 1α,25-Dihydroxyvitamin D3 reduces several types of UV-induced DNA damage and contributes to photoprotection. J. Steroid Biochem. Mol. Biol. 2013, 136, 131–138. [Google Scholar] [CrossRef]
- Kim, J.S.; Jung, M.; Yoo, J.; Choi, E.H.; Park, B.C.; Kim, M.H.; Hong, S.P. Protective Effect of Topical Vitamin D(3) against Photocarcinogenesis in a Murine Model. Ann. Dermatol. 2016, 28, 304–313. [Google Scholar] [CrossRef]
- Geldenhuys, S.; Hart, P.H.; Endersby, R.; Jacoby, P.; Feelisch, M.; Weller, R.B.; Matthews, V.; Gorman, S. Ultraviolet Radiation Suppresses Obesity and Symptoms of Metabolic Syndrome Independently of Vitamin D in Mice Fed a High-Fat Diet. Diabetes 2014, 63, 3759–3769. [Google Scholar] [CrossRef]
- Weller, R.B. The health benefits of UV radiation exposure through vitamin D production or non-vitamin D pathways. Blood pressure and cardiovascular disease. Photochem. Photobiol. Sci. 2017, 16, 374–380. [Google Scholar] [CrossRef]
- Teng, S.; Chakravorty, L.; Fleury, N.; Gorman, S. Regular exposure to non-burning ultraviolet radiation reduces signs of non-alcoholic fatty liver disease in mature adult mice fed a high fat diet: Results of a pilot study. BMC Res. Notes 2019, 12, 78. [Google Scholar] [CrossRef]
- Havinga, E.; Bots, J.P.L. Studies on vitamin D, I. the synthesis of vitamin D3 3 C14. Recl. Trav. Chim. Pays-Bas 1954, 73, 393–400. [Google Scholar] [CrossRef]
- Heilbron, I.M.; Spring, F.S.; Stewart, P.A. 289. Studies in the sterol group. Part XXI. Lumisterol. J. Chem. Soc. 1935, 1221–1223. [Google Scholar] [CrossRef]
- Gottfried, N.; Kaiser, W.; Braun, M.; Fuss, W.; Kompa, K.L. Ultrafast electrocyclic ring opening in previtamin D photochemistry. Chem. Phys. Lett. 1984, 110, 335–339. [Google Scholar] [CrossRef]
- Jacobs, H.J.C.; Havinga, E. Photochemistry of Vitamin D and its Isomers and of Simple Trienes. In Advances in Photochemistry; Pitts, J.N., Jr., Hammond, G.S., Gollnick, K., Grosjean, D., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1979; Volume 11, pp. 305–373. [Google Scholar]
- Omura, T.; Sato, R. A New Cytochrome in Liver Microsomes. J. Biol. Chem. 1962, 237, PC1375–PC1376. [Google Scholar] [CrossRef]
- Skobowiat, C.; Dowdy, J.C.; Sayre, R.M.; Tuckey, R.C.; Slominski, A. Cutaneous hypothalamic-pituitary-adrenal axis homolog: Regulation by ultraviolet radiation. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E484–E493. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.-K.; Li, W.; Yi, A.-K.; Postlethwaite, A.; Tuckey, R.C. The role of CYP11A1 in the production of vitamin D metabolites and their role in the regulation of epidermal functions. J. Steroid Biochem. Mol. Biol. 2014, 144, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Tuckey, R.C.; Slominski, A.T.; Cheng, C.Y.S.; Chen, J.; Kim, T.-K.; Xiao, M.; Li, W. Lumisterol is metabolized by CYP11A1: Discovery of a new pathway. Int. J. Biochem. Cell Biol. 2014, 55, 24–34. [Google Scholar] [CrossRef]
- Tuckey, R.C.; Li, W.; Ma, D.; Cheng, C.Y.S.; Wang, K.M.; Kim, T.K.; Jeayeng, S.; Slominski, A.T. CYP27A1 acts on the pre-vitamin D3 photoproduct, lumisterol, producing biologically active hydroxy-metabolites. J. Steroid Biochem. Mol. Biol. 2018, 181, 1–10. [Google Scholar] [CrossRef]
- Tongkao-on, W.; Carter, S.; Reeve, V.E.; Dixon, K.M.; Gordon-Thomson, C.; Halliday, G.M.; Tuckey, R.C.; Mason, R.S. CYP11A1 in skin: An alternative route to photoprotection by vitamin D compounds. J. Steroid Biochem. Mol. Biol. 2015, 148, 72–78. [Google Scholar] [CrossRef]
- Chaiprasongsuk, A.; Janjetovic, Z.; Kim, T.-K.; Schwartz, C.J.; Tuckey, R.; Tang, E.; Raman, C.; Panich, U.; Slominski, A. Hydroxylumisterols, Photoproducts of Pre-Vitamin D3, Protect Human Keratinocytes against UVB-Induced Damage. Int. J. Mol. Sci. 2020, 21, 9374. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.-K.; Hobrath, J.V.; Janjetovic, Z.; Oak, A.S.W.; Postlethwaite, A.; Lin, Z.; Li, W.; Takeda, Y.; Jetten, A.M.; et al. Characterization of a new pathway that activates lumisterol in vivo to biologically active hydroxylumisterols. Sci. Rep. 2017, 7, 11434. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Li, W.; Kim, T.-K.; Semak, I.; Wang, J.; Zjawiony, J.K.; Tuckey, R.C. Novel activities of CYP11A1 and their potential physiological significance. J. Steroid Biochem. Mol. Biol. 2015, 151, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Tuckey, R.C.; Tang, E.K.Y.; Chen, Y.A.; Slominski, A.T. Selective ability of rat 7-Dehydrocholesterol reductase (DHCR7) to act on some 7-Dehydrocholesterol metabolites but not on lumisterol metabolites. J. Steroid Biochem. Mol. Biol. 2021, 212, 105929. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, N.S.; Greenoak, G.E.; Mason, R.S. Effects of ultraviolet irradiation on human skin-derived epidermal cells in vitro. J. Cell Physiol. 1993, 157, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, V.B.; Rybchyn, M.S.; Gordon-Thomson, C.; Tongkao-on, W.; Mizwicki, M.T.; Norman, A.W.; Reeve, V.E.; Halliday, G.M.; Mason, R.S. Opening of Chloride Channels by 1[alpha],25-Dihydroxyvitamin D3 Contributes to Photoprotection against UVR-Induced Thymine Dimers in Keratinocytes. J. Investig. Derm. 2013, 133, 776–782. [Google Scholar] [CrossRef]
- Aasen, T.; Izpisua Belmonte, J.C. Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nat. Protoc. 2010, 5, 371–382. [Google Scholar] [CrossRef]
- McLeod, S.D.; Smith, C.; Mason, R.S. Stimulation of tyrosinase in human melanocytes by pro-opiomelanocortin-derived peptides. J. Endocrinol. 1995, 146, 439–447. [Google Scholar] [CrossRef]
- Rybchyn, M.S.; De Silva, W.G.M.; Sequeira, V.B.; McCarthy, B.Y.; Dilley, A.V.; Dixon, K.M.; Halliday, G.M.; Mason, R.S. Enhanced repair of UV-induced DNA damage by 1,25-Dihydroxyvitamin D3 in skin is linked to pathways that control cellular energy. J. Investig. Dermatol. 2018, 138, 1146–1156. [Google Scholar] [CrossRef]
- Tuckey, R.C.; Li, W.; Zjawiony, J.K.; Zmijewski, M.A.; Nguyen, M.N.; Sweatman, T.; Miller, D.; Slominski, A. Pathways and products for the metabolism of vitamin D3 by cytochrome P450scc. FEBS J. 2008, 275, 2585–2596. [Google Scholar] [CrossRef]
- Cooke, M.S.; Robson, A. Immunochemical detection of UV-induced DNA damage and repair. Methods Mol. Biol. 2006, 314, 215–228. [Google Scholar] [CrossRef]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.; Gupta, R.; Dixon, K.M.; Deo, S.S.; Choong, S.M.; Halliday, G.M.; Bishop, J.E.; Ishizuka, S.; Norman, A.W.; Posner, G.H.; et al. 1,25-Dihydroxyvitamin D and three low-calcemic analogs decrease UV-induced DNA damage via the rapid response pathway. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Miura, T.; Furuichi, M.; Tominaga, Y.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006, 16, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, V.B.; Rybchyn, M.S.; Tongkao-On, W.; Gordon-Thomson, C.; Malloy, P.J.; Nemere, I.; Norman, A.W.; Reeve, V.E.; Halliday, G.M.; Feldman, D.; et al. The role of the vitamin D receptor and ERp57 in photoprotection by 1alpha,25-dihydroxyvitamin D3. Mol. Endocrinol. 2012, 26, 574–582. [Google Scholar] [CrossRef]
- Hönigsmann, H.; Brenner, W.; Tanew, A.; Ortel, B. UV-Induced unscheduled DNA synthesis in human skin: Dose response, correlation with erythema, time course and split dose exposure in vivo. J. Photochem. Photobiol. B Biol. 1987, 1, 33–43. [Google Scholar] [CrossRef]
- Pieper, A.A.; Verma, A.; Zhang, J.; Snyder, S.H. Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol. Sci. 1999, 20, 171–181. [Google Scholar] [CrossRef]
- Tettamanti, G.; Malagoli, D.; Ottaviani, E.; De Eguileor, M. Oligomycin A and the IPLB-LdFB insect cell line: Actin and mitochondrial responses. Cell Biol. Int. 2008, 32, 287–292. [Google Scholar] [CrossRef]
- Tettamanti, G.; Malagoli, D.; Marchesini, E.; Congiu, T.; De Eguileor, M.; Ottaviani, E. Oligomycin A induces autophagy in the IPLB-LdFB insect cell line. Cell Tissue Res. 2006, 326, 179–186. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313 Pt 1, 17–29. [Google Scholar] [CrossRef]
- Rybchyn, M.S.; Brennan-Speranza, T.C.; Mor, D.; Cheng, Z.; Chang, W.; Conigrave, A.D.; Mason, R.S. The mTORC2 Regulator Homer1 Modulates Protein Levels and Sub-Cellular Localization of the CaSR in Osteoblast-Lineage Cells. Int. J. Mol. Sci. 2021, 22, 6509. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.M.; Deo, S.S.; Norman, A.W.; Bishop, J.E.; Halliday, G.M.; Reeve, V.E.; Mason, R.S. In vivo relevance for photoprotection by the vitamin D rapid response pathway. J. Steroid Biochem. Mol. Biol. 2007, 103, 451–456. [Google Scholar] [CrossRef] [PubMed]
- De Haes, P.; Garmyn, M.; Verstuyf, A.; De Clercq, P.; Vandewalle, M.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. 1,25-Dihydroxyvitamin D3 and analogues protect primary human keratinocytes against UVB-induced DNA damage. J. Photochem. Photobiol. B Biol. 2005, 78, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Janjetovic, Z.; Kim, T.-K.; Wasilewski, P.; Rosas, S.; Hanna, S.; Sayre, R.M.; Dowdy, J.C.; Li, W.; Tuckey, R.C. Novel non-calcemic secosteroids that are produced by human epidermal keratinocytes protect against solar radiation. J. Steroid Biochem. Mol. Biol. 2015, 148, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Beyer, R.E. The effect of ultraviolet light on mitochondria. I. Inactivation and protection of oxidative phosphorylation during far-ultraviolet irradiation. Arch. Biochem. Biophys. 1959, 79, 269–274. [Google Scholar] [CrossRef]
- Maglio, D.H.G.; Paz, M.L.; Ferrari, A.; Weill, F.S.; Czerniczyniec, A.; Leoni, J.; Bustamante, J. Skin damage and mitochondrial dysfunction after acute ultraviolet B irradiation: Relationship with nitric oxide production. Photodermatol. Photoimmunol. Photomed. 2005, 21, 311–317. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Gezen-Ak, D.; Alaylıoğlu, M.; Yurttaş, Z.; Çamoğlu, T.; Şengül, B.; İşler, C.; Kına, Ü.Y.; Keskin, E.; Atasoy, İ.L.; Kafardar, A.M.; et al. Vitamin D receptor regulates transcription of mitochondrial DNA and directly interacts with mitochondrial DNA and TFAM. J. Nutr. Biochem. 2023, 116, 109322. [Google Scholar] [CrossRef] [PubMed]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Nagai, Y.; Sladek, R.; Bastien, Y.; Ho, J.; Petrecca, K.; Sotiropoulou, G.; Diamandis, E.P.; Hudson, T.J.; White, J.H. Expression profiling in squamous carcinoma cells reveals pleiotropic effects of vitamin D3 analog EB1089 signaling on cell proliferation, differentiation, and immune system regulation. Mol. Endocrinol. 2002, 16, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am. J. Hypertens. 2014, 27, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D cell signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71. [Google Scholar] [CrossRef]
- Chaiprasongsuk, A.; Janjetovic, Z.; Kim, T.-K.; Holick, M.F.; Tuckey, R.C.; Panich, U.; Slominski, A.T. 274—Protective effects of novel derivatives of vitamin D3 and lumisterol against UVB-induced damage in human keratinocytes involve activation of Nrf2 and P53 defense mechanisms. Free Radic. Biol. Med. 2019, 128, S116. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.-K.; Slominski, R.M.; Song, Y.; Janjetovic, Z.; Podgorska, E.; Reddy, S.B.; Song, Y.; Raman, C.; Tang, E.K.Y.; et al. Metabolic activation of tachysterol3 to biologically active hydroxyderivatives that act on VDR, AhR, LXRs, and PPARγ receptors. FASEB J. 2022, 36, e22451. [Google Scholar] [CrossRef]
- Burney, S.; Caulfield, J.L.; Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1999, 424, 37–49. [Google Scholar] [CrossRef]
- Jaiswal, M.; LaRusso, N.F.; Burgart, L.J.; Gores, G.J. Inflammatory Cytokines Induce DNA damage and Inhibit DNA repair in Cholangiocarcinoma Cells by a Nitric Oxide-dependent Mechanism. Cancer Res. 2000, 60, 184–190. [Google Scholar]
- Steeg, H.V.; Vries, A.D.; Oostrom, C.T.M.V.; Benthem, J.V.; Beems, R.B.; Kreijl, C.F.V. DNA Repair—Deficient Xpa and Xpa/p53+/− Knock-Out Mice: Nature of the Models. Toxicol. Pathol. 2001, 29, 109–116. [Google Scholar] [CrossRef]
- Volker, M.; Moné, M.J.; Karmakar, P.; Van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.J.; Van Driel, R.; Van Zeeland, A.A.; Mullenders, L.H.F. Sequential Assembly of the Nucleotide Excision Repair Factors In Vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-E.; Zhu, Q.; Wani, G.; El-Mahdy, M.A.; Li, J.; Wani, A.A. DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res. 2005, 33, 4023–4034. [Google Scholar] [CrossRef] [PubMed]
- Moll, P.R.; Sander, V.; Frischauf, A.-M.; Richter, K. Expression profiling of vitamin D treated primary human keratinocytes. J. Cell. Biochem. 2007, 100, 574–592. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.Y. Photoprotection by 1(alpha),25-Dihydroxyvitamin D3 and Vitamin D Like Compound in Human Skin. Master’s Thesis, University of Sydney, Sydney, Australia, 2014. [Google Scholar]
- Leslie, N.R.; Batty, I.H.; Maccario, H.; Davidson, L.; Downes, C.P. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene 2008, 27, 5464–5476. [Google Scholar] [CrossRef]
- Ming, M.; Han, W.; Maddox, J.; Soltani, K.; Shea, C.R.; Freeman, D.M.; He, Y.Y. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene 2010, 29, 492–502. [Google Scholar] [CrossRef]
- Ming, M.; Feng, L.; Shea, C.R.; Soltani, K.; Zhao, B.; Han, W.; Smart, R.C.; Trempus, C.S.; He, Y.Y. PTEN positively regulates UVB-induced DNA damage repair. Cancer Res. 2011, 71, 5287–5295. [Google Scholar] [CrossRef]
- Shariev, A. Novel targets for Vitamin D in the Inhibition of Melanoma Growth and Metastasis. Ph.D. Thesis, University of Sydney, Sydney Medical School, Sydney, Australia, 2019. [Google Scholar]
- Shariev, A.; Painter, N.; Reeve, V.E.; Haass, N.K.; Rybchyn, M.S.; Ince, F.A.; Mason, R.S.; Dixon, K.M. PTEN: A novel target for vitamin D in melanoma. J. Steroid Biochem. Mol. Biol. 2022, 218, 106059. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.K.; Qayyum, S.; Song, Y.; Janjetovic, Z.; Oak, A.S.W.; Slominski, R.M.; Raman, C.; Stefan, J.; Mier-Aguilar, C.A.; et al. Vitamin D and lumisterol derivatives can act on liver X receptors (LXRs). Sci. Rep. 2021, 11, 8002. [Google Scholar] [CrossRef]
- De Silva, W.G.M.; Han, J.Z.R.; Yang, C.; Tongkao-On, W.; McCarthy, B.Y.; Ince, F.A.; Holland, A.J.A.; Tuckey, R.C.; Slominski, A.T.; Abboud, M.; et al. Evidence for Involvement of Nonclassical Pathways in the Protection From UV-Induced DNA Damage by Vitamin D–Related Compounds. JBMR Plus 2021, 5, e10555. [Google Scholar] [CrossRef]
- Ellison, T.I.; Smith, M.K.; Gilliam, A.C.; MacDonald, P.N. Inactivation of the Vitamin D Receptor Enhances Susceptibility of Murine Skin to UV-induced Tumorigenesis. J. Investig. Dermatol. 2008, 128, 2508–2517. [Google Scholar] [CrossRef]
- Zinser, G.M.; Sundberg, J.P.; Welsh, J. Vitamin D3 receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis 2002, 23, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.J.; Segaert, S.; Degreef, H.; Bouillon, R.; Garmyn, M. Ultraviolet B Suppresses Vitamin D Receptor Gene Expression in Keratinocytes. Biochem. Biophys. Res. Commun. 1998, 246, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Chaiprasongsuk, A.; Janjetovic, Z.; Kim, T.K.; Stefan, J.; Slominski, R.M.; Hanumanthu, V.S.; Raman, C.; Qayyum, S.; Song, Y.; et al. Photoprotective Properties of Vitamin D and Lumisterol Hydroxyderivatives. Cell Biochem. Biophys. 2020, 78, 165–180. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.K.; Li, W.; Postlethwaite, A.; Tieu, E.W.; Tang, E.K.Y.; Tuckey, R.C. Detection of novel CYP11A1-derived secosteroids in the human epidermis and serum and pig adrenal gland. Sci. Rep. 2015, 5, 14875. [Google Scholar] [CrossRef] [PubMed]
- Tongkao-on, W. Role of Vitamin D and Other Compounds in the Protection of Skin Cells from UV. Ph.D. Thesis, University of Sydney, Sydney, Australia, 2015. [Google Scholar]
- Teichert, A.E.; Elalieh, H.; Elias, P.M.; Welsh, J.; Bikle, D.D. Overexpression of hedgehog signaling is associated with epidermal tumor formation in vitamin D receptor-null mice. J. Investig. Dermatol. 2011, 131, 2289–2297. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Silva, W.G.M.; McCarthy, B.Y.; Han, J.; Yang, C.; Holland, A.J.A.; Stern, H.; Dixon, K.M.; Tang, E.K.Y.; Tuckey, R.C.; Rybchyn, M.S.; et al. The Over-Irradiation Metabolite Derivative, 24-Hydroxylumister-ol3, Reduces UV-Induced Damage in Skin. Metabolites 2023, 13, 775. https://doi.org/10.3390/metabo13070775
De Silva WGM, McCarthy BY, Han J, Yang C, Holland AJA, Stern H, Dixon KM, Tang EKY, Tuckey RC, Rybchyn MS, et al. The Over-Irradiation Metabolite Derivative, 24-Hydroxylumister-ol3, Reduces UV-Induced Damage in Skin. Metabolites. 2023; 13(7):775. https://doi.org/10.3390/metabo13070775
Chicago/Turabian StyleDe Silva, Warusavithana Gunawardena Manori, Bianca Yuko McCarthy, Jeremy Han, Chen Yang, Andrew J. A. Holland, Harvey Stern, Katie Marie Dixon, Edith Kai Yan Tang, Robert Charles Tuckey, Mark Stephen Rybchyn, and et al. 2023. "The Over-Irradiation Metabolite Derivative, 24-Hydroxylumister-ol3, Reduces UV-Induced Damage in Skin" Metabolites 13, no. 7: 775. https://doi.org/10.3390/metabo13070775