

Cancer Metabolism: The Role of ROS in DNA Damage and Induction of Apoptosis in Cancer Cells

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. ROS Levels in Cancer Cells and Cancer Development
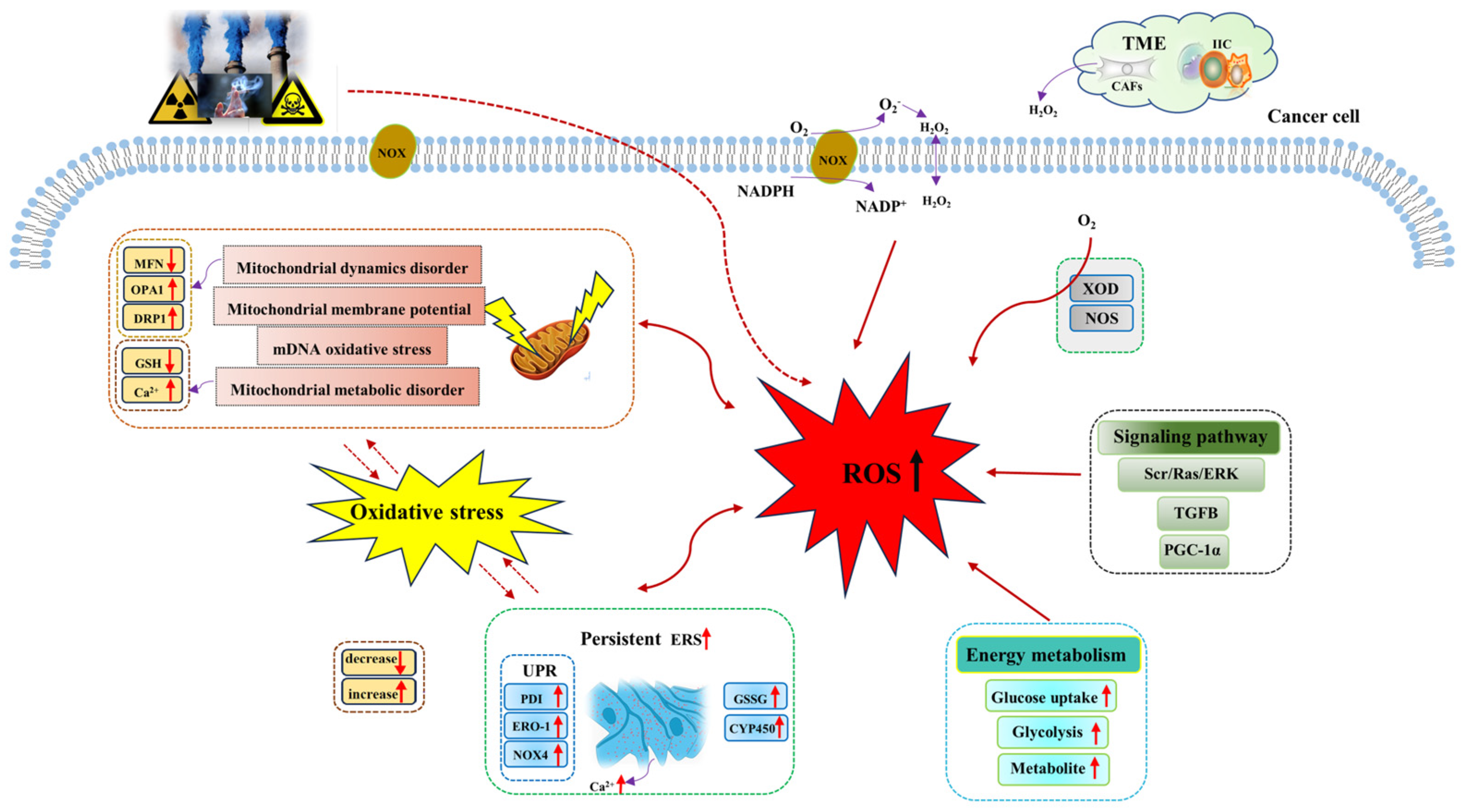
2.1. Mechanisms of ROS Generation in Cancer
2.2. The Dual Role of ROS in Promoting and Suppressing Cancer Cells
2.2.1. High Levels of Steady-State ROS Play a Role in Promoting Cancer
2.2.2. Toxic Levels of ROS Play a Pro-Apoptotic Role
2.3. Changes in the Adaptation of Cancer Cells to ROS
3. Cancer Therapeutic Strategies Targeting ROS Generation
3.1. Drug Therapies That Target ROS Generation
3.2. Novel Therapies Targeting ROS Generation
3.3. Pro-Oxidative Anticancer Agents Targeting ROS Generation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hernandez, D.; Cheng, C.Y.; Hernandez-Villafuerte, K.; Schlander, M. Survival and comorbidities in lung cancer patients: Evidence from administrative claims data in Germany. Oncol. Res. 2022, 30, 173–185. [Google Scholar] [CrossRef]
- Ezzati, M.; Riboli, E. Behavioral and dietary risk factors for noncommunicable diseases. N. Engl. J. Med. 2013, 369, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Aune, D.; Chan, D.S.M.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Fedewa, S.A.; Anderson, W.F.; Miller, K.D.; Ma, J.; Rosenberg, P.S.; Jemal, A. Colorectal cancer incidence patterns in the United States, 1974-2013. J. Natl. Cancer Inst. 2017, 109, djw322. [Google Scholar] [CrossRef] [Green Version]
- Araghi, M.; Soerjomataram, I.; Jenkins, M.; Brierley, J.; Morris, E.; Bray, F.; Arnold, M. Global trends in colorectal cancer mortality: Projections to the year 2035. Int. J. Cancer 2019, 144, 2992–3000. [Google Scholar] [CrossRef] [Green Version]
- Lucky, S.S.; Soo, K.C.; Zhang, Y. Nanoparticles in photodynamic therapy. Chem. Rev. 2015, 115, 1990–2042. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Bergamini, C.; Leoni, I.; Rizzardi, N.; Melli, M.; Galvani, G.; Coada, C.A.; Giovannini, C.; Monti, E.; Liparulo, I.; Valenti, F.; et al. MiR-494 induces metabolic changes through G6pc targeting and modulates sorafenib response in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2023, 42, 145. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.; Fang, Y.; Yao, C.; Wang, Y.; Zhang, Z.; Wang, X.; Gao, J.; Feng, Y.; Sun, L.; Zou, R.; et al. The synergistic effects of PRDX5 and Nrf2 on lung cancer progression and drug resistance under oxidative stress in the zebrafish models. Oncol. Res. 2022, 30, 53–64. [Google Scholar] [CrossRef]
- Ebata, H.; Loo, T.M.; Takahashi, A. Telomere maintenance and the cGAS-STING pathway in cancer. Cells 2022, 11, 1958. [Google Scholar] [CrossRef]
- Morin, G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 1989, 59, 521–529. [Google Scholar] [CrossRef]
- Cuadrado, M.; Martinez-Pastor, B.; Fernandez-Capetillo, O. ATR activation in response to ionizing radiation: Still ATM territory. Cell Div. 2006, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, P.J. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu. Rev. Pathol. 2012, 7, 303–321. [Google Scholar] [CrossRef] [Green Version]
- Huiting, W.; Dekker, S.L.; van der Lienden, J.C.J.; Mergener, R.; Musskopf, M.K.; Furtado, G.V.; Gerrits, E.; Coit, D.; Oghbaie, M.; Di Stefano, L.H.; et al. Targeting DNA topoisomerases or checkpoint kinases results in an overload of chaperone systems, triggering aggregation of a metastable subproteome. eLife 2022, 11, e70726. [Google Scholar] [CrossRef]
- Priya, B.; Ravi, S.; Kirubakaran, S. Targeting ATM and ATR for cancer therapeutics: Inhibitors in clinic. Drug Discov. Today 2023, 28, 103662. [Google Scholar] [CrossRef]
- Kavec, M.J.; Urbanova, M.; Makovicky, P.; Opattová, A.; Tomasova, K.; Kroupa, M.; Kostovcikova, K.; Siskova, A.; Navvabi, N.; Schneiderova, M.; et al. Oxidative damage in sporadic colorectal cancer: Molecular mapping of base excision repair glycosylases MUTYH and hOGG1 in colorectal cancer patients. Int. J. Mol. Sci. 2022, 23, 5704. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Shu, P.; Liang, H.; Zhang, J.; Lin, Y.; Chen, W.; Zhang, D. Reactive oxygen species formation and its effect on CD4(+) T cell-mediated inflammation. Front. Immunol. 2023, 14, 1199233. [Google Scholar] [CrossRef]
- Chen, Y.J.; Guo, X.; Liu, M.L.; Yu, Y.Y.; Cui, Y.H.; Shen, X.Z.; Liu, T.S.; Liang, L. Interaction between glycolysis-cholesterol synthesis axis and tumor microenvironment reveal that gamma-glutamyl hydrolase suppresses glycolysis in colon cancer. Front. Immunol. 2022, 13, 979521. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [Green Version]
- Sinha, B.K.; Tokar, E.J.; Bortner, C.D. Molecular mechanisms of cytotoxicity of NCX4040, the non-steroidal anti-inflammatory NO-donor, in human ovarian cancer cells. Int. J. Mol. Sci. 2022, 23, 8611. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, B.K.; Tokar, E.J.; Li, J.; Bushel, P.R. Gene expression profiling elucidates cellular responses to NCX4040 in human ovarian tumor cells: Implications in the mechanisms of action of NCX4040. Cancers 2023, 15, 285. [Google Scholar] [CrossRef] [PubMed]
- Popovici, V.; Musuc, A.M.; Matei, E.; Karampelas, O.; Ozon, E.A.; Cozaru, G.C.; Schröder, V.; Bucur, L.; Aricov, L.; Anastasescu, M.; et al. ROS-induced DNA-damage and autophagy in oral squamous cell carcinoma by Usnea barbata oil extract-an in vitro study. Int. J. Mol. Sci. 2022, 23, 14836. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Rogoff, H.A. Implications of reactive oxygen species on cancer formation and its treatment. Semin. Oncol. Cancer 2021, 48, 238–245. [Google Scholar] [CrossRef]
- Valavanidis, A.; Fiotakis, K.; Vlachogianni, T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Health Pt. C-Environ. Carcinog. Ecotoxicol. Rev. 2008, 26, 339–362. [Google Scholar] [CrossRef]
- Liu, F.; Xu, T.; Ng, N.L.; Lu, H. Linking cell health and reactive oxygen species from secondary organic aerosols exposure. Environ. Sci. Technol. 2023, 57, 1039–1048. [Google Scholar] [CrossRef]
- Niture, S.; Gadi, S.; Lin, M.; Qi, Q.; Niture, S.S.; Moore, J.T.; Bodnar, W.; Fernando, R.A.; Levine, K.E.; Kumar, D. Cadmium modulates steatosis, fibrosis, and oncogenic signaling in liver cancer cells by activating notch and AKT/mTOR pathways. Environ. Toxicol. 2023, 38, 783–797. [Google Scholar] [CrossRef]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef] [Green Version]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [Green Version]
- Xiang, D.; Yang, W.; Fang, Z.; Mao, J.; Yan, Q.; Li, L.; Tan, J.; Yu, C.; Qian, J.; Tang, D.; et al. Agrimol B inhibits colon carcinoma progression by blocking mitochondrial function through the PGC-1α/NRF1/TFAM signaling pathway. Front. Oncol. 2022, 12, 1055126. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; She, W.; Li, Y.; Wang, M.; Liu, Y.; Ning, B.; Xu, T.; Huang, T.; Wei, Y. Aa-Z2 triggers ROS-induced apoptosis of osteosarcoma by targeting PDK-1. J. Transl. Med. 2023, 21, 7. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sierra, T.; Jiménez-Uribe, A.P.; Ortega-Lozano, A.J.; Ramírez-Magaña, K.J.; Pedraza-Chaverri, J. Antioxidants affect endoplasmic reticulum stress-related diseases. In Vitamins and Hormones: Antioxidants, 1st ed.; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA; Elsevier Inc.: Oxford, UK, 2023; Volume 121, pp. 169–196. [Google Scholar] [CrossRef]
- Yang, L.; Li, A.; Wang, Y.; Zhang, Y. Intratumoral microbiota: Roles in cancer initiation, development and therapeutic efficacy. Signal Transduct. Target. Ther. 2023, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Romesberg, A.; Van Houten, B. Targeting mitochondrial function with chemoptogenetics. Biomedicines 2022, 10, 2459. [Google Scholar] [CrossRef]
- Sylvester, A.L.; Zhang, D.X.; Ran, S.; Zinkevich, N.S. Inhibiting NADPH oxidases to target vascular and other pathologies: An update on recent experimental and clinical studies. Biomolecules 2022, 12, 823. [Google Scholar] [CrossRef]
- Bánfi, B.; Clark, R.A.; Steger, K.; Krause, K.H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 2003, 278, 3510–3513. [Google Scholar] [CrossRef] [Green Version]
- Juhasz, A.; Markel, S.; Gaur, S.; Liu, H.; Lu, J.; Jiang, G.; Wu, X.; Antony, S.; Wu, Y.; Melillo, G.; et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 2017, 292, 7866–7887. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.S.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; Li, L.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2018, 8, e2562. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, L.; Moldovan, N.I. Oxygen free radicals and redox biology of organelles. Histochem. Cell Biol. 2004, 122, 395–412. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Denisenko, T.V.; Gorbunova, A.S.; Zhivotovsky, B. Mitochondrial involvement in migration, invasion and metastasis. Front. Cell Dev. Biol. 2019, 7, 355. [Google Scholar] [CrossRef]
- Yan, X.; Liang, F.; Li, D.; Zheng, J. Ouabain elicits human glioblastoma cells apoptosis by generating reactive oxygen species in ERK-p66SHC-dependent pathway. Mol. Cell Biochem. 2015, 398, 95–104. [Google Scholar] [CrossRef]
- Liu-Smith, F.; Dellinger, R.; Meyskens, F.L., Jr. Updates of reactive oxygen species in melanoma etiology and progression. Arch. Biochem. Biophys. 2014, 563, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, H.; Liu, C.; Yuan, X. Role of ROS-mediated autophagy in melanoma (Review). Mol. Med. Rep. 2022, 26, 303. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.Z.; Nisar, M.A.; Alshwmi, M.; Din, S.R.U.; Gamallat, Y.; Khan, M.; Ma, T. Brevilin A inhibits STAT3 signaling and induces ROS-dependent apoptosis, mitochondrial stress and endoplasmic reticulum stress in MCF-7 breast cancer cells. Onco Targets Ther. 2020, 13, 435–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldi, S.; He, Y.; Ivanov, I.; Sun, Y.; Feng, W.; Refat, M.; Mohammed, S.A.D.; Adlat, S.; Tian, Z.; Wang, Y.; et al. Novel characterization discoveries of ferroptosis-associated molecules in COAD microenvironment based TCGA data. Front. Mol. Biosci. 2022, 9, 1102735. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Swaroop, S.; Dutta, N.; Arya, A.; Ghosh, S.; Dhabal, S.; Das, P.; Majumder, C.; Pal, M.; Bhattacharjee, A. IL-13 and the hydroperoxy fatty acid 13(S)HpODE play crucial role in inducing an apoptotic pathway in cancer cells involving MAO-A/ROS/p53/p21 signaling axis. Free Radic. Biol. Med. 2023, 195, 309–328. [Google Scholar] [CrossRef]
- Kwon, M.; Jung, J.; Park, H.S.; Kim, N.H.; Lee, J.; Park, J.; Kim, Y.; Shin, S.; Lee, B.S.; Cheong, Y.H.; et al. Diesel exhaust particle exposure accelerates oxidative DNA damage and cytotoxicity in normal human bronchial epithelial cells through PD-L1. Environ. Pollut. 2023, 317, 120705. [Google Scholar] [CrossRef]
- Gong, G.; Ganesan, K.; Xiong, Q.; Zheng, Y. Antitumor effects of ononin by modulation of apoptosis in non-small-cell lung cancer through inhibiting PI3K/Akt/mTOR pathway. Oxid. Med. Cell Longev. 2022, 2022, 5122448. [Google Scholar] [CrossRef]
- Polyakov, N.; Leshina, T.; Fedenok, L.; Slepneva, I.; Kirilyuk, I.; Furso, J.; Olchawa, M.; Sarna, T.; Elas, M.; Bilkis, I.; et al. Redox-active quinone chelators: Properties, mechanisms of action, cell delivery, and cell toxicity. Antioxid. Redox Signal. 2018, 28, 1394–1403. [Google Scholar] [CrossRef]
- Huang, Y.F.; Zhu, D.J.; Chen, X.W.; Chen, Q.K.; Luo, Z.T.; Liu, C.C.; Wang, G.X.; Zhang, W.J.; Liao, N.Z. Curcumin enhances the effects of irinotecan on colorectal cancer cells through the generation of reactive oxygen species and activation of the endoplasmic reticulum stress pathway. Oncotarget 2017, 8, 40264–40275. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, M.; Zou, P.; Kanchana, K.; Weng, Q.; Chen, W.; Zhong, P.; Ji, J.; Zhou, H.; He, L.; et al. Curcumin analog WZ35 induced cell death via ROS-dependent ER stress and G2/M cell cycle arrest in human prostate cancer cells. BMC Cancer 2015, 15, 866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Chen, J.; Zeng, T.; Huang, Q.; Chen, D.; Chen, H.; Chen, J.; Zheng, B.; Wang, M.; Chen, S.; et al. WZ35 inhibits gastric cancer cell metastasis by depleting glutathione to promote cellular metabolic remodeling. Cancer Lett. 2023, 555, 216044. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, T.H.; Choi, W.G.; Chung, Y.H.; Ko, S.G.; Cheon, C.; Cho, S.G. SH003 causes ER stress-mediated apoptosis of breast cancer cells via intracellular ROS production. Cancer Genom. Proteom. 2023, 20, 88–116. [Google Scholar] [CrossRef]
- Si, Z.; Yang, G.; Wang, X.; Yu, Z.; Pang, Q.; Zhang, S.; Qian, L.; Ruan, Y.; Huang, J.; Yu, L. An unconventional cancer-promoting function of methamphetamine in hepatocellular carcinoma. Life Sci. Alliance 2023, 6, e202201660. [Google Scholar] [CrossRef]
- Woo, J.H.; Seo, H.J.; Lee, J.Y.; Lee, I.; Jeon, K.; Kim, B.; Lee, K. Polypropylene nanoplastic exposure leads to lung inflammation through p38-mediated NF-κB pathway due to mitochondrial damage. Part. Fibre Toxicol. 2023, 20, 2. [Google Scholar] [CrossRef]
- Benavides, R.A.S.; Leiro-Vidal, J.M.; Rodriguez-Gonzalez, J.A.; Ares-Pena, F.J.; López-Martín, E. The HL-60 human promyelocytic cell line constitutes an effective in vitro model for evaluating toxicity, oxidative stress and necrosis/apoptosis after exposure to black carbon particles and 2.45 GHz radio frequency. Sci. Total Environ. 2023, 867, 161475. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Meng, F.; Sui, C.; Jiang, Y.; Zhang, L. Arsenic enhances cell death and DNA damage induced by ultraviolet B exposure in mouse epidermal cells through the production of reactive oxygen species. Clin. Exp. Dermatol. 2019, 44, 512–519. [Google Scholar] [CrossRef]
- Giuntini, F.; Foglietta, F.; Marucco, A.M.; Troia, A.; Dezhkunov, N.V.; Pozzoli, A.; Durando, G.; Fenoglio, I.; Serpe, L.; Canaparo, R. Insight into ultrasound-mediated reactive oxygen species generation by various metal-porphyrin complexes. Free Radic. Biol. Med. 2018, 121, 190–201. [Google Scholar] [CrossRef]
- Lv, W.; Sui, L.; Yan, X.; Xie, H.; Jiang, L.; Geng, C.; Li, Q.; Yao, X.; Kong, Y.; Cao, J. ROS-dependent Atg4 upregulation mediated autophagy plays an important role in Cd-induced proliferation and invasion in A549 cells. Chem. Biol. Interact. 2018, 279, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Zinflou, C.; Rochette, P.J. Absorption of blue light by cigarette smoke components is highly toxic for retinal pigmented epithelial cells. Arch. Toxicol. 2019, 93, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Yang, Y.; Guo, C.; Zhang, R.; Sun, S.; Wang, Y.; Qiao, Q.; Fu, Y.; Pang, Q. NOX4-derived ROS mediates TGF-β1-induced metabolic reprogramming during epithelial-mesenchymal transition through the PI3K/AKT/HIF-1α pathway in glioblastoma. Oxid. Med. Cell Longev. 2021, 2021, 5549047. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Chandrasekaran, B.; Navin, A.K.; Shukla, V.; Baby, B.V.; Ankem, M.K.; Damodaran, C. Molecular interplay between NOX1 and autophagy in cadmium-induced prostate carcinogenesis. Free Radic. Biol. Med. 2023, 199, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Zhu, H.; Ma, H.; Dong, N.; Wu, M.; Li, D.; Liu, L.; Shi, Q.; Ju, X. 1,5-anhydroglucitol promotes pre-B acute lymphocytic leukemia progression by driving glycolysis and reactive oxygen species formation. BMC Cancer 2023, 23, 122. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Q.; Zhang, Z.; Ji, M.; Du, T.; Jin, J.; Jiang, J.D.; Chen, X.; Hu, H.Y. Characterization of chlorogenic acid as a two-photon fluorogenic probe that regulates glycolysis in tumor cells under hypoxia. J. Med. Chem. 2023, 66, 2498–2505. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Lindahl, T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 2004, 38, 445–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, C.J.; Muller, J.G. Oxidative nucleobase modifications leading to strand scission. Chem. Rev. 1998, 98, 1109–1152. [Google Scholar] [CrossRef]
- Li, C.; Xue, Y.; Ba, X.; Wang, R. The role of 8-oxoG repair systems in tumorigenesis and cancer therapy. Cells 2022, 11, 3798. [Google Scholar] [CrossRef]
- Boiteux, S.; Coste, F.; Castaing, B. Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: Properties and biological roles of the Fpg and OGG1 DNA N-glycosylases. Free Radic. Biol. Med. 2017, 107, 179–201. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Zhao, R.; Sun, F.; Lu, Q.; Wang, X.; Hu, J.; Wang, S.; Gao, L.; Zhou, Q.; Xiong, X.; et al. TXNDC9 regulates oxidative stress-induced androgen receptor signaling to promote prostate cancer progression. Oncogene 2020, 39, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.H.; Huang, P.H.; Hsu, Y.J.; Peng, Y.J.; Lee, C.H.; Wang, J.C.; Chen, J.W.; Lin, S.J. Inhibition of hypoxia inducible factor-1α attenuates abdominal aortic aneurysm progression through the down-regulation of matrix metalloproteinases. Sci. Rep. 2016, 6, 28612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cao, Y.; Guo, X.; Wang, X.; Han, X.; Kanwore, K.; Hong, X.; Zhou, H.; Gao, D. Hypoxia-induced ROS aggravate tumor progression through HIF-1α-SERPINE1 signaling in glioblastoma. J. Zhejiang Univ.-SCI. B 2023, 24, 32–49. [Google Scholar] [CrossRef]
- Gao, Y.; Nan, X.; Shi, X.; Mu, X.; Liu, B.; Zhu, H.; Yao, B.; Liu, X.; Yang, T.; Hu, Y.; et al. SREBP1 promotes the invasion of colorectal cancer accompanied upregulation of MMP7 expression and NF-κB pathway activation. BMC Cancer 2019, 19, 685. [Google Scholar] [CrossRef] [Green Version]
- Eller-Borges, R.; Rodrigues, E.G.; Teodoro, A.C.S.; Moraes, M.S.; Arruda, D.C.; Paschoalin, T.; Curcio, M.F.; da Costa, P.E.; Do Nascimento, I.R.; Calixto, L.A.; et al. Bradykinin promotes murine melanoma cell migration and invasion through endogenous production of superoxide and nitric oxide. Nitric Oxide-Biol. Chem. 2023, 132, 15–26. [Google Scholar] [CrossRef]
- Robinson, A.J.; Hopkins, G.L.; Rastogi, N.; Hodges, M.; Doyle, M.; Davies, S.; Hole, P.S.; Omidvar, N.; Darley, R.L.; Tonks, A. Reactive oxygen species drive proliferation in acute myeloid leukemia via the glycolytic regulator PFKFB3. Cancer Res. 2020, 80, 937–949. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Rowe, L.A.; Degtyareva, N.; Doetsch, P.W. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2008, 45, 1167–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Ma, Y.; Liu, K.; Gao, J.; Li, S.; Sun, X.; Li, G. Resveratrol induces DNA damage-mediated cancer cell senescence through the DLC1-DYRK1A-EGFR axis. Food Funct. 2023, 14, 1484–1497. [Google Scholar] [CrossRef]
- Yu, T.T.; Hu, J.; Li, Q.R.; Peng, X.C.; Xu, H.Z.; Han, N.; Li, L.G.; Yang, X.X.; Xu, X.; Yang, Z.Y.; et al. Chlorin e6-induced photodynamic effect facilitates immunogenic cell death of lung cancer as a result of oxidative endoplasmic reticulum stress and DNA damage. Int. Immunopharmacol. 2023, 115, 109661. [Google Scholar] [CrossRef]
- Arjmand, F.; Yasir Khan, H.; Tabassum, S. Progress of metal-based anticancer chemotherapeutic agents in last two decades and their comprehensive biological (DNA/RNA binding, cleavage and cytotoxicity activity) studies. Chem. Rec. 2023, 23, e202200247. [Google Scholar] [CrossRef]
- Carmody, R.J.; Cotter, T.G. Signalling apoptosis: A radical approach. Redox Rep. 2001, 6, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, S.H.; Shang, X.J.; Yu, L.S.; Zhu, J.W.; Zhao, A.; Zhou, Y.F.; An, G.H.; Zhang, Q.; Ma, B. Triptolide induces Sertoli cell apoptosis in mice via ROS/JNK-dependent activation of the mitochondrial pathway and inhibition of Nrf2-mediated antioxidant response. Acta Pharmacol. Sin. 2018, 39, 311–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, N.J.; Davis, R.J. Role of JNK in tumor development. Cell Cycle 2003, 2, 199–201. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, S.; Han, K.; Wang, L.; Liu, X. Induction of apoptosis by matrine derivative ZS17 in human hepatocellular carcinoma BEL-7402 and HepG2 cells through ROS-JNK-P53 signalling pathway activation. Int. J. Mol. Sci. 2022, 23, 15991. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xu, G.; Xu, Q.; Xu, C.; Zhou, X.; Bai, Y.; Yin, L.; Ding, Y.; Wang, W. MLN2238 exerts its anti-tumor effects via regulating ROS/JNK/mitochondrial signaling pathways in intrahepatic cholangiocarcinoma. Front. Pharmacol. 2022, 13, 1040847. [Google Scholar] [CrossRef]
- Lv, Y.; Du, Y.; Li, K.; Ma, X.; Wang, J.; Du, T.; Ma, Y.; Teng, Y.; Tang, W.; Ma, R.; et al. The FACT-targeted drug CBL0137 enhances the effects of rituximab to inhibit B-cell non-Hodgkin’s lymphoma tumor growth by promoting apoptosis and autophagy. Cell Commun. Signal. 2023, 21, 16. [Google Scholar] [CrossRef]
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox Signal. 2009, 11, 2655–2671. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Hohorst, L.; John, M.; Albert, M.C.; King, L.E.; Beckmann, L.; Szabo, T.; Hertlein, V.; Luo, X.; Villunger, A.; et al. BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. EMBO J. 2022, 41, e108690. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, T.; Maryam, A.; Tian, X.; Khan, M.; Ma, T. Santamarine inhibits NF-кB and STAT3 activation and induces apoptosis in HepG2 liver cancer cells via oxidative stress. J. Cancer 2017, 8, 3707–3717. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Yan, Y.; Qu, J.; Xue, X.; Liu, Z.; Cai, H. Emodin induces apoptosis of lung cancer cells through ER stress and the TRIB3/NF-κB pathway. Oncol. Rep. 2017, 37, 1565–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhang, J.; Guo, G.; Cai, Y.; Cui, R.; Yin, C.; Liu, W.; Vinothkumar, R.; Zhang, T.; Liang, G.; et al. A mono-carbonyl analog of curcumin induces apoptosis in drug-resistant EGFR-mutant lung cancer through the generation of oxidative stress and mitochondrial dysfunction. Cancer Manag. Res. 2018, 10, 3069–3082. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, P.; Wallace, A.; D’Armiento, J.M. Induction of the unfolded protein response by cigarette smoke is primarily an activating transcription factor 4-C/EBP homologous protein mediated process. Int. J. Chron. Obstruct. Pulmon. Dis. 2011, 6, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [Green Version]
- Piao, M.J.; Han, X.; Kang, K.A.; Fernando, P.D.S.M.; Herath, H.M.U.L.; Hyun, J.W. The endoplasmic reticulum stress response mediates shikonin-induced apoptosis of 5-fluorouracil-resistant colorectal cancer cells. Biomol. Ther. 2022, 30, 265–273. [Google Scholar] [CrossRef]
- Chen, W.; Zou, P.; Zhao, Z.; Weng, Q.; Chen, X.; Ying, S.; Ye, Q.; Wang, Z.; Ji, J.; Liang, G. Selective killing of gastric cancer cells by a small molecule via targeting TrxR1 and ROS-mediated ER stress activation. Oncotarget 2016, 7, 16593–16609. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Yu, C.I.; Lee, T.H.; Chuang, J.M.; Han, K.F.; Lin, C.S.; Huang, W.P.; Chen, J.Y.; Chen, C.Y.; Lin, M.Y.; et al. Plumbagin induces the apoptosis of drug-resistant oral cancer in vitro and in vivo through ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction. Phytomedicine 2023, 111, 154655. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Chen, H.; Liang, J.; Li, Y.; Yang, J.; Luo, C.; Tang, Y.; Ding, Y.; Liu, X.; Yuan, Q.; et al. Dual role of reactive oxygen species and their application in cancer therapy. J. Cancer 2021, 12, 5543–5561. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Egashira, A.; Igarashi, H.; Iwakuma, T.; Nakatsuru, Y.; Tominaga, Y.; Kawate, H.; Nakao, K.; Nakamura, K.; Ide, F.; et al. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. Natl. Acad. Sci. USA 2001, 98, 11456–11461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakehashi, A.; Ishii, N.; Okuno, T.; Fujioka, M.; Gi, M.; Wanibuchi, H. Enhanced susceptibility of Ogg1 mutant mice to multiorgan carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1801. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.M.; Chiang, J.C.; Shang, Z.; Palchik, G.; Newman, C.; Zhang, Y.; Davis, A.J.; Lee, H.; Chen, B.P. DNA-PKcs and ATM modulate mitochondrial ADP-ATP exchange as an oxidative stress checkpoint mechanism. EMBO J. 2023, 42, e112094. [Google Scholar] [CrossRef]
- Luo, M.; Wicha, M.S. Targeting cancer stem cell redox metabolism to enhance therapy responses. Semin. Radiat. Oncol. 2019, 29, 42–54. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslanbaeva, L.R.; Santoro, M.M. Adaptive redox homeostasis in cutaneous melanoma. Redox Biol. 2020, 37, 101753. [Google Scholar] [CrossRef] [PubMed]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The role of Nrf2 activity in cancer development and progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [Green Version]
- Robertson, H.; Dinkova-Kostova, A.T.; Hayes, J.D. NRF2 and the ambiguous consequences of its activation during initiation and the subsequent stages of tumourigenesis. Cancers 2020, 12, 3609. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yu, L.; Xu, C.; Li, Y.M.; Zhao, Y.R.; Cao, M.M.; Yang, L.Y. NAD(P)HX epimerase downregulation promotes tumor progression through ROS/HIF-1α signaling in hepatocellular carcinoma. Cancer Sci. 2021, 112, 2753–2769. [Google Scholar] [CrossRef]
- Willson, J.A.; Arienti, S.; Sadiku, P.; Reyes, L.; Coelho, P.; Morrison, T.; Rinaldi, G.; Dockrell, D.H.; Whyte, M.K.B.; Walmsley, S.R. Neutrophil HIF-1α stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood 2022, 139, 281–286. [Google Scholar] [CrossRef]
- Boakye, D.; Jansen, L.; Halama, N.; Chang-Claude, J.; Hoffmeister, M.; Brenner, H. Early discontinuation and dose reduction of adjuvant chemotherapy in stage III colon cancer patients. Ther. Adv. Med. Oncol. 2021, 13, 1–18. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, C.; Zhou, F.; Zhang, Y. Shikonin potentiates therapeutic efficacy of oxaliplatin through reactive oxygen species-mediated intrinsic apoptosis and endoplasmic reticulum stress in oxaliplatin-resistant colorectal cancer cells. Drug Dev. Res. 2023, 84, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Truong Hoang, Q.; Huynh, K.A.; Nguyen Cao, T.G.; Kang, J.H.; Dang, X.N.; Ravichandran, V.; Kang, H.C.; Lee, M.; Kim, J.E.; Ko, Y.T.; et al. Piezocatalytic 2D WS2 nanosheets for ultrasound-triggered and mitochondria-targeted piezodynamic cancer therapy synergized with energy metabolism-targeted chemotherapy. Adv. Mater. 2023, 35, 2300437. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Kiang, K.M.; Cheng, S.Y.; Leung, G.K. Pharmacological inhibition of serine synthesis enhances temozolomide efficacy by decreasing O6-methylguanine DNA methyltransferase (MGMT) expression and reactive oxygen species (ROS)-mediated DNA damage in glioblastoma. Lab. Investig. 2022, 102, 194–203. [Google Scholar] [CrossRef]
- Atashi, F.; Vahed, N.; Emamverdizadeh, P.; Fattahi, S.; Paya, L. Drug resistance against 5-fluorouracil and cisplatin in the treatment of head and neck squamous cell carcinoma: A systematic review. J. Dent. Res. Dent. Clin. Dent. Prospect. 2021, 15, 219–225. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Moreira, H.; Szyjka, A.; Paliszkiewicz, K.; Barg, E. Prooxidative activity of celastrol induces apoptosis, DNA damage, and cell cycle arrest in drug-resistant human colon cancer cells. Oxid. Med. Cell Longev. 2019, 2019, 6793957. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, J.; Chen, Q.; Song, H.; Wang, Z.; Xing, W.; Jin, S.; Song, X.; Yang, H.; Zhao, W. The gut microbiota metabolite urolithin B prevents colorectal carcinogenesis by remodeling microbiota and PD-L1/HLA-B. Oxid. Med. Cell Longev. 2023, 2023, 6480848. [Google Scholar] [CrossRef]
- Kawiak, A.; Domachowska, A.; Lojkowska, E. Plumbagin increases paclitaxel-induced cell death and overcomes paclitaxel resistance in breast cancer cells through ERK-mediated apoptosis induction. J. Nat. Prod. 2019, 82, 878–885. [Google Scholar] [CrossRef]
- Choi, J.A.; Lee, E.H.; Cho, H.; Kim, J.H. High-dose selenium induces ferroptotic cell death in ovarian cancer. Int. J. Mol. Sci. 2023, 24, 1918. [Google Scholar] [CrossRef]
- Gawel, A.M.; Singh, R.; Debinski, W. Metal-based nanostructured therapeutic strategies for glioblastoma treatment—An update. Biomedicines 2022, 10, 1598. [Google Scholar] [CrossRef]
- Yokoi, K.; Yasuda, Y.; Kanbe, A.; Imura, T.; Aoki, S. Development of wireless power-transmission-based photodynamic therapy for the induction of cell death in cancer cells by cyclometalated iridium(III) complexes. Molecules 2023, 28, 1433. [Google Scholar] [CrossRef]
- Nkune, N.W.; Kruger, C.A.; Abrahamse, H. Synthesis of a novel nanobioconjugate for targeted photodynamic therapy of colon cancer enhanced with cannabidiol. Oncotarget 2022, 13, 156–172. [Google Scholar] [CrossRef]
- Crous, A.; Abrahamse, H. Effective gold nanoparticle-antibody-mediated drug delivery for photodynamic therapy of lung cancer stem cells. Int. J. Mol. Sci. 2020, 21, 3742. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, W.; Luo, N.; Xue, Z.; Hu, Q.; Zeng, W.; Xu, J. Bimetallic nanocrystals: Structure, controllable synthesis and applications in catalysis, energy and sensing. Nanomaterials 2021, 11, 1926. [Google Scholar] [CrossRef]
- Jiang, W.; Liang, M.; Lei, Q.; Li, G.; Wu, S. The current status of photodynamic therapy in cancer treatment. Cancers 2023, 15, 585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yi, H.; Song, J.; Huang, J.; Yang, K.; Tan, B.; Wang, D.; Yang, N.; Wang, Z.; Li, X. Mitochondria-targeted and ultrasound-activated nanodroplets for enhanced deep-penetration sonodynamic cancer therapy. ACS Appl. Mater. Interfaces 2019, 11, 9355–9366. [Google Scholar] [CrossRef]
- Greco, G.; Ulfo, L.; Turrini, E.; Marconi, A.; Costantini, P.E.; Marforio, T.D.; Mattioli, E.J.; Di Giosia, M.; Danielli, A.; Fimognari, C.; et al. Light-enhanced cytotoxicity of doxorubicin by photoactivation. Cells 2023, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Wang, F.; Liu, Y.; Zhou, J.; Zhao, W.; Lu, L.; Li, J.; He, X. Dual-cascade activatable nanopotentiators reshaping adenosine metabolism for sono-chemodynamic-immunotherapy of deep tumors. Adv. Sci. 2023, 10, 2207200. [Google Scholar] [CrossRef]
- Zhu, B.; Zhang, M.; Chen, Q.; Li, Z.; Chen, S.; Zhu, J. Starvation-assisted and photothermal-thriving combined chemo/chemodynamic cancer therapy with PT/MR bimodal imaging. Biomater. Sci. 2023, 11, 2129–2138. [Google Scholar] [CrossRef]
- Chen, Y.; Li, J.; Zhao, Z. Redox control in acute lymphoblastic leukemia: From physiology to pathology and therapeutic opportunities. Cells 2021, 10, 1218. [Google Scholar] [CrossRef]
- Khan, M.; Maryam, A.; Saleem, M.Z.; Shakir, H.A.; Qazi, J.I.; Li, Y.; Ma, T. Brevilin A induces ROS-dependent apoptosis and suppresses STAT3 activation by direct binding in human lung cancer cells. J. Cancer 2020, 11, 3725–3735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravanakumar, G.; Kim, J.; Kim, W.J. Reactive-oxygen-species-responsive drug delivery systems: Promises and challenges. Adv. Sci. 2017, 4, 1600124. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cui, H.; Li, M.; Chai, Z.; Wang, H.; Jin, X.; Dai, F.; Liu, Y.; Zhou, B. Tumor killing by a dietary curcumin mono-carbonyl analog that works as a selective ROS generator via TrxR inhibition. Eur. J. Med. Chem. 2023, 250, 115191. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.R.; Singh, R.N.; Carroll, D.L.; Wood, J.C.; D’Agostino, R.B., Jr.; Ajayan, P.M.; Torti, F.M.; Torti, S.V. The resistance of breast cancer stem cells to conventional hyperthermia and their sensitivity to nanoparticle-mediated photothermal therapy. Biomaterials 2012, 33, 2961–2970. [Google Scholar] [CrossRef] [Green Version]
- Najafabad, B.K.; Attaran, N.; Mahmoudi, M.; Sazgarnia, A. Effect of photothermal and photodynamic therapy with cobalt ferrite superparamagnetic nanoparticles loaded with LCG and PpIX on cancer stem cells in MDA-MB-231 and A375 cell lines. Photodiagnosis Photodyn. Ther. 2023, 43, 103648. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Chirila, R.M.; Dronca, R.S. Immune checkpoint inhibitor toxicities. Mayo Clin. Proc. 2019, 94, 1321–1329. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Cao, X.; Shen, B.; Chen, Z.; Chen, G. Injectable cold atmospheric plasma-activated immunotherapeutic hydrogel for enhanced cancer treatment. Biomaterials 2023, 300, 122189. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.Y.; Chang, K.F.; Huang, Y.C.; Huang, X.F.; Sheu, G.T.; Kuo, C.F.; Hsiao, C.Y.; Tsai, N.M. Patchouli alcohol induces G(0)/G(1) cell cycle arrest and apoptosis in vincristine-resistant non-small cell lung cancer through ROS-mediated DNA damage. Thorac. Cancer 2023, 1–11. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, X.; Ou, X.; Lee, M.M.S.; Zhang, J.; Xu, C.; Yu, X.; Gong, P.; Lam, J.W.Y.; Zhang, P.; et al. Tailoring the amphiphilic structure of zwitterionic AIE photosensitizers to boost antitumor immunity. Adv. Mater. 2023, e2303186. [Google Scholar] [CrossRef]
- Ma, X.; Zhou, W.; Zhang, R.; Zhang, C.; Yan, J.; Feng, J.; Rosenholm, J.M.; Shi, T.; Shen, X.; Zhang, H. Minimally invasive injection of biomimetic Nano@Microgel for in situ ovarian cancer treatment through enhanced photodynamic reactions and photothermal combined therapy. Mater. Today Bio 2023, 20, 100663. [Google Scholar] [CrossRef] [PubMed]
- Aniogo, E.C.; George, B.P.; Abrahamse, H. Molecular effectors of photodynamic therapy-mediated resistance to cancer cells. Int. J. Mol. Sci. 2021, 22, 13182. [Google Scholar] [CrossRef]
- Saini, H.; Sharma, H.; Mukherjee, S.; Chowdhury, S.; Chowdhury, R. Verteporfin disrupts multiple steps of autophagy and regulates p53 to sensitize osteosarcoma cells. Cancer Cell Int. 2021, 21, 52. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Objective | Aim | Results | Reference |
|---|---|---|---|
| Air pollutants | To demonstrate that oxidative stress is the key mechanism by which secondary organic aerosols affect cell health. | To a large extent, cellular health depends on cellular ROS levels, and secondary organic aerosols exposure produce biological effects through oxidative stress. | [37] |
| To investigate mechanism of oxidative stress and cytotoxicity induced by DEPs exposure in HBE. | Cells treated with DEPs showed high levels of ROS generation and oxidative DNA damage. | [60] | |
| Anticancer drugs | To investigate whether ononin synergizes with paclitaxel to inhibit non-small-cell lung cancer progression and promote apoptosis in vitro and in vivo. | The combination of ononin and paclitaxel increased the expression of ROS generation and apoptotic markers and inhibited cell proliferation through the PI3K/Akt/mTOR signaling pathways. | [61] |
| To investigate mechanism of action regarding redox-active quinone chelators. | Redox-active quinone chelators can be reduced by ETC and GSH while generating ROS. They can also generate ROS upon photoexcitation. | [62] | |
| To explore the anticancer mechanism of Brv-A. | Brv-A induces ROS generation; however, Brv-A regulates ROS generation through NOX2 and NOX3 proteins and the induction of ER-stress in MCF-7 BC cells. | [57] | |
| To investigate the antitumor mechanism of Agr. | Agr increased ROS, blocked Bcl-2 expression, and increased Caspase-3 and Bax expression to promote apoptosis in cancer cells. | [41] | |
| To investigate the effect of curcumin on the effect of irinotecan on CRC cells. | Curcumin enhances the killing effect of irinotecan on CRC cells by mediating increased ROS production and the activation of the ER stress pathway. | [63] | |
| To determine the effect and mechanism of action of curcumin analog WZ35 against human prostate cancer. | WZ35 exhibited strong antitumor potential against PC-3 cells by inducing ROS generation and subsequently inducing ER stress-dependent apoptosis and cell cycle arrest. | [64] | |
| To reveal the mechanism of WZ35-mediated ROS generation and amino acid metabolism regulation that inhibit gastric cancer cell metastasis. | WZ35 can deplete the GSH reserve by increasing ROS generation. The mechanism maintains the GSH consumption phenotype through the ROS-YAP-AXL-ALKBH5-GLS2 loop. | [65] | |
| To investigate the molecular mechanism of human BC cells apoptosis caused by SH003. | SH003 induced apoptosis in BC cells by increasing ROS generation and activating the ER stress signaling pathways. | [66] | |
| Chemicals | To explore the key mechanism of methamphetamine in HCC. | ROS-mediated Ras up-regulation activates the MEK/ERK signaling pathway, which is key mechanism by which methamphetamine promotes HCC progress. | [67] |
| To investigate the antitumor mechanism of organic arsenic compound Aa-Z2 on osteosarcoma by targeting cancer metabolism. | Aa-Z2 induces ROS accumulation by targeting PDK-1 to induce osteosarcoma apoptosis. | [42] | |
| To explore the mechanism of mitochondrial damage after exposure to toxic polypropylene nanoplastic. | Polypropylene nanoplastic stimulation leads to mitochondrial dysfunction and ROS generation and causes lung inflammation through the p38-mediated NF-κB pathway. | [68] | |
| Radiation | To evaluate the combined action of EMFs and black carbon particles in the HL-60 promyelocytic cell line exposed to 2.45 GHz RF radiation. | The interaction between black carbon particles and RF leads to ROS generation and triggers oxidative stress to activate necrosis/apoptosis, leading to long-term cytotoxicity. | [69] |
| Heavy metals | To study the co-carcinogenic effects of UVB and arsenic on mouse epidermal cell line JB6 and its mechanism. | Arsenic enhances UVB-induced ROS generation and causes DNA damage and apoptosis in mouse skin cells. | [70] |
| To investigate the ROS generation and related bio-effects of various metal-porphyrin complexes under ultrasonic exposure. | Zn(II) and Pd(II) porphyrins are the most effective in producing singlet oxygen and hydroxyl radicals, and the different patterns of ROS generation depend on the metal moiety. | [71] | |
| To investigate the role of ROS- associated autophagy in Cd-induced cell proliferation and the invasion of A549 cells. | Exposure to Cd (2 μM) significantly increased ROS accumulation, induced autophagy, and enhanced cell growth, migration, and invasion in A549 cells. | [72] | |
| To investigate the effects of low concentrations of Cd on the regulation of liver cancer cell proliferation, steatosis, and fibrogenic/ oncogenic signaling. | Exposure to Cd (1–10 nM) increases ROS production, cell proliferation, steatosis, and fibrogenic/oncogenic signaling by activating Notch and AKT/mTOR pathways. | [38] | |
| Cigarette smoke | To assess the toxic effects of PAH and HEV light combination in human RPE cells. | Toxic synergistic interaction between IcdP and HEV light. This synergy translates into the disruption of the mitochondrial network, enhanced ROS accumulation, and apoptosis. | [73] |
| Cytokine | To investigate the role of IL-13 and 13(S)HpODE (endogenous product during IL-13 activation) in mediating apoptotic pathways in three different in vitro cellular models: A549 lung cancer, HCT116 colorectal cancer, and CCF52 GBM cells. | 13(S)HpODE significantly reduces solid tumor growth through the activation of apoptosis. IL-13 and 13(S)HpODE participate in activating the p53-p21 signaling cascade via MAO-A-mediated ROS and ultimately induce apoptosis by inhibiting Bcl-2 and promoting Bax. | [59] |
| FRGs | To explore and verify the mechanism by which FRGs promotes the progression and invasion of colon adenocarcinoma. | FRGs improves tumor cell survival by activating the TGFB pathway, which can stimulate ROS generation, accelerate ECM decomposition, and promote tumor progression and invasion. | [58] |
| Mitochondria damage | To investigate the molecular mechanism of ouabain-induced ROS generation and apoptosis in human GBM cells. | Ouabain-induced GBM cells apoptosis increased ROS generation through the phosphorylation of p66Shc (mediated by the Src/Ras/ERK signaling pathways). | [54] |
| NOX | To investigate in GBM cell lines that cause TGF-β1 to drive metabolic reprogramming and aggressive cancer by enhancing NOX4 activity. | TGF-β1 up-regulated NOX4 expression accompanied by ROS through Smad-dependent signaling and then induced HIF-1α overexpression and metabolic reprogramming while promoting EMT (modulated by the PI3K/AKT/HIF-1α signaling pathways). | [74] |
| To investigate the role of NOX complex proteins in the Cd-induced malignant transformation of prostate epithelial cells and the molecular mechanisms involved. | Chronic Cd exposure activated NOX1 complex proteins and generated ROS and ER stress, which led to defective autophagy. | [75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Ye, X.; Xiong, Z.; Ihsan, A.; Ares, I.; Martínez, M.; Lopez-Torres, B.; Martínez-Larrañaga, M.-R.; Anadón, A.; Wang, X.; et al. Cancer Metabolism: The Role of ROS in DNA Damage and Induction of Apoptosis in Cancer Cells. Metabolites 2023, 13, 796. https://doi.org/10.3390/metabo13070796
Zhao Y, Ye X, Xiong Z, Ihsan A, Ares I, Martínez M, Lopez-Torres B, Martínez-Larrañaga M-R, Anadón A, Wang X, et al. Cancer Metabolism: The Role of ROS in DNA Damage and Induction of Apoptosis in Cancer Cells. Metabolites. 2023; 13(7):796. https://doi.org/10.3390/metabo13070796
Chicago/Turabian StyleZhao, Yongxia, Xiaochun Ye, Zhifeng Xiong, Awais Ihsan, Irma Ares, Marta Martínez, Bernardo Lopez-Torres, María-Rosa Martínez-Larrañaga, Arturo Anadón, Xu Wang, and et al. 2023. "Cancer Metabolism: The Role of ROS in DNA Damage and Induction of Apoptosis in Cancer Cells" Metabolites 13, no. 7: 796. https://doi.org/10.3390/metabo13070796
APA StyleZhao, Y., Ye, X., Xiong, Z., Ihsan, A., Ares, I., Martínez, M., Lopez-Torres, B., Martínez-Larrañaga, M.-R., Anadón, A., Wang, X., & Martínez, M.-A. (2023). Cancer Metabolism: The Role of ROS in DNA Damage and Induction of Apoptosis in Cancer Cells. Metabolites, 13(7), 796. https://doi.org/10.3390/metabo13070796