
Iron Absorption: Molecular and Pathophysiological Aspects



Abstract
:1. Introduction
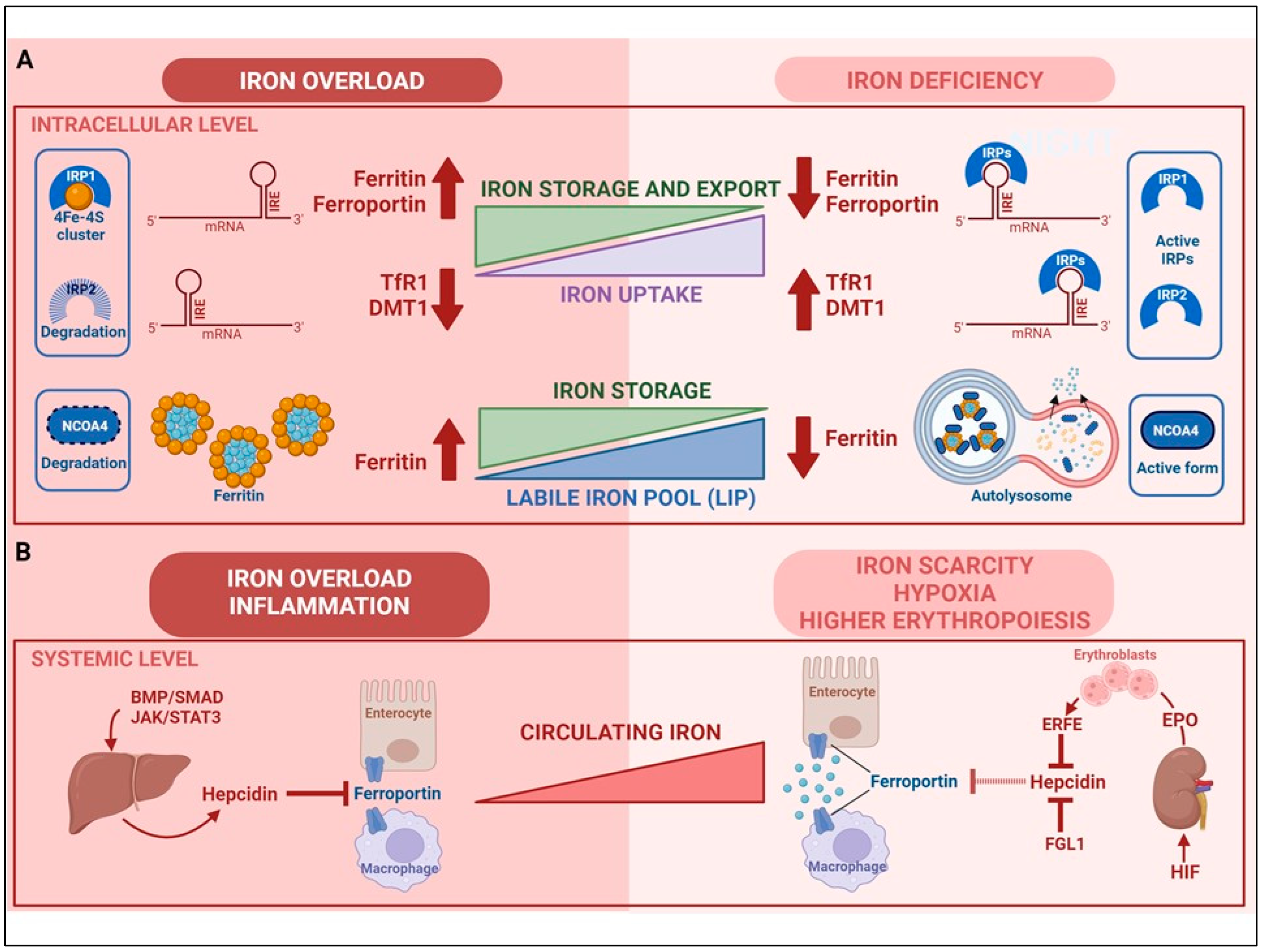
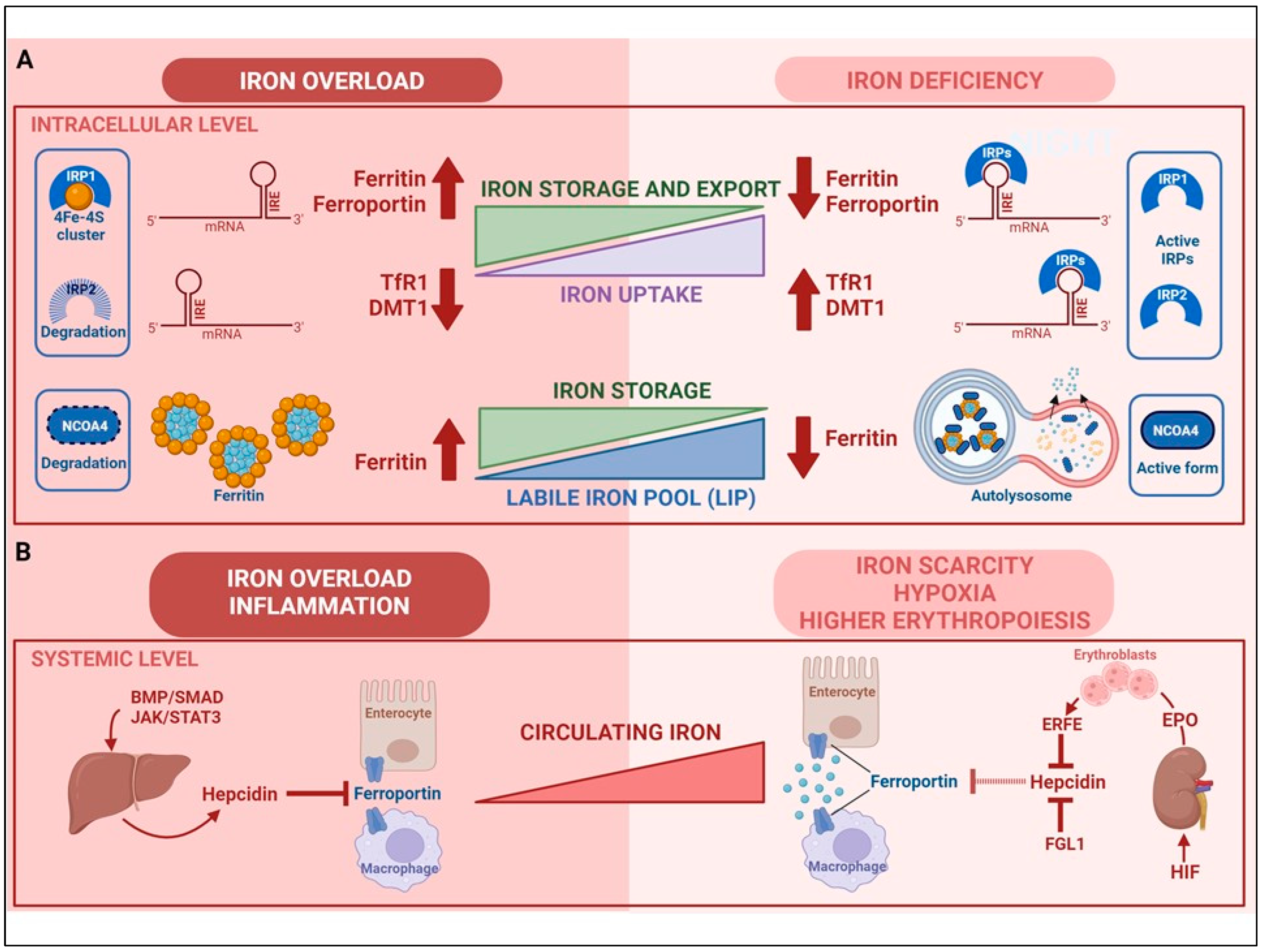
2. Regulation of Cellular Iron Homeostasis
3. Regulation of Systemic Iron Homeostasis
4. Iron Absorption
Mechanisms of Iron Absorption
- (a)
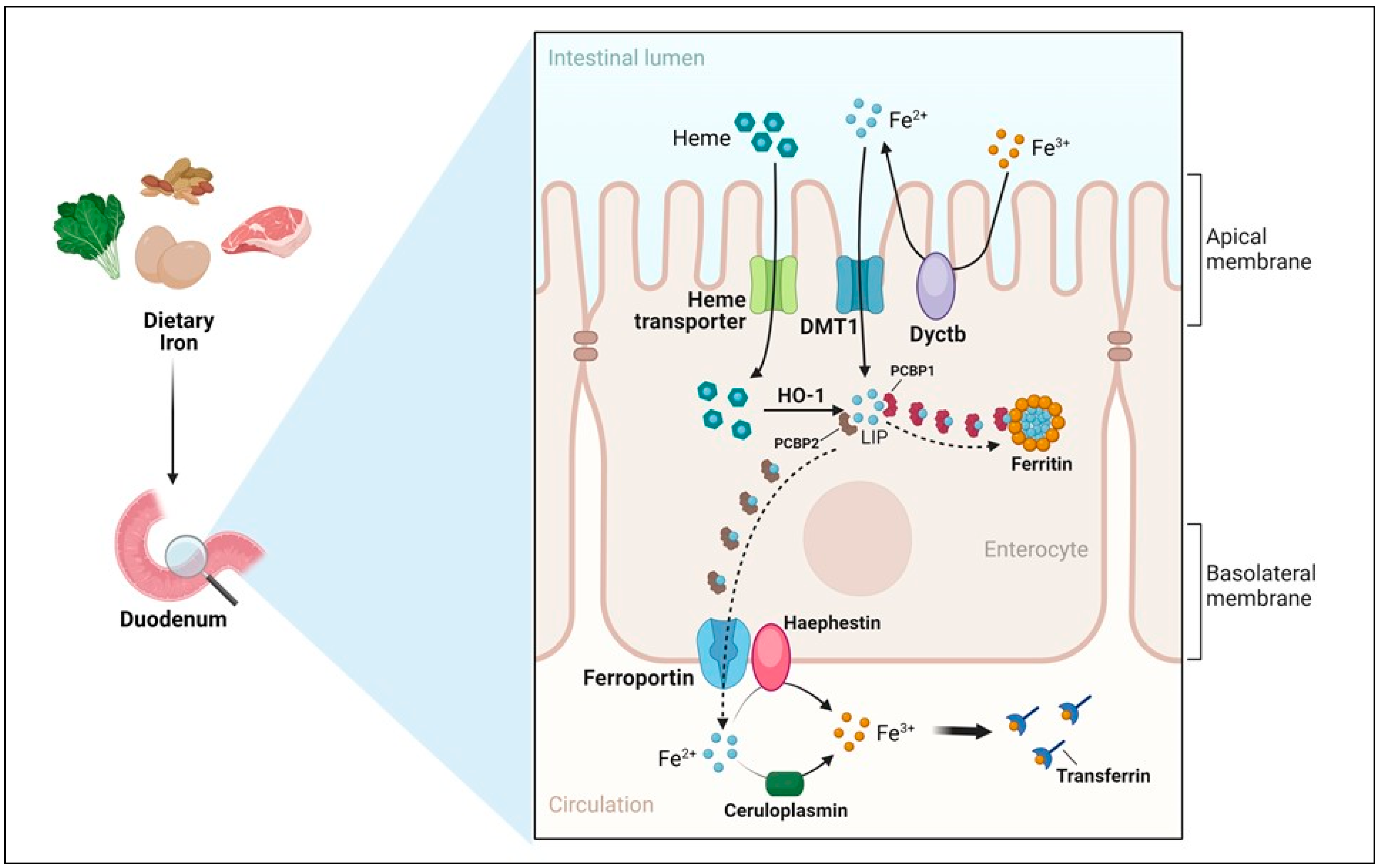
- Apical uptake. The uptake of nutritional nonheme iron, which has been elucidated at the molecular level, occurs mostly in the first portion of the duodenum and involves the transport of Fe2+ across the apical membrane of enterocytes by DMT1 (Figure 2). However, since Fe3+ is the form of iron mostly present in the diet and the low pH present in the intestinal lumen is not sufficient to maintain iron in a soluble form, the previous reduction of iron by ferric reductases, such as Dcytb (duodenal cytochrome b) is required [15]. Accordingly, the importance of diet composition in determining the amount of iron absorbed is well recognized. Indeed, the assumption of reductants like vitamin C can improve iron absorption by making the task of Dcytb easier. Conversely, food components mainly present in vegetables like phytates, which are primarily found in cereals and legumes, or tannins, may reduce iron absorption because of unspecific binding, physical entrapment and decreased intestinal transit time. The strategy aimed at achieving better iron bioavailability by decreasing the consumption of food containing these inhibitors should be matched against the recent and warranted trend toward increasing the intake of insoluble fiber. However, phytases to remove phytic acid from food are increasingly used in food-processing techniques to reduce these inhibiting effects. Other nutrients, including minerals like calcium and vitamins, could possibly impair iron absorption (reviewed in [48]). Dietary heme, originating primarily from meat and seafood, can also be transported across the apical membrane by a hitherto poorly known mechanism. In fact, heme carrier protein 1 (HCP1), which was initially identified as an intestinal heme importer, turned out to transport folate, for which HCP1 has an affinity much higher than for heme. Alternatively, heme responsive gene (HRG1) that transports heme across the erythrophagosomal membranes of macrophages during iron recycling from RBC [49] and is expressed in the human small intestine, could represent a candidate for intestinal heme absorption, but its role in this context is still unknown [44]. In any case, it is well established that dietary absorbed heme is subsequently catabolized within intestinal epithelial cells by heme oxygenase 1 (HO-1) to liberate Fe2+, which then follows the same destiny of inorganic iron imported by DMT1.
- (b)
- Enterocytic intracellular phase. Internalized Fe2+ enters the LIP in the enterocytic cytoplasm and, as in any other cell, is either utilized, incorporated in ferritin, or exported by ferroportin at the basolateral surface (see below) (Figure 2). Given the function of the duodenum in body iron absorption, the latter fate is predominant. Recently, a key role for the chaperone PCBP1 in intestinal iron absorption has been reported [50]; the cell-specific deletion of PCBP1 in mice led to lower iron and ferritin levels in enterocytes and disrupted iron balance. As already mentioned, iron not used by the duodenal cells is either reversibly stored in ferritin or exported by ferroportin. Whether ferritin levels simply reflect the iron status of the enterocyte or play an active role in the control of absorption has been long discussed. Indeed, we found that in line with the corresponding IRP binding activity, ferritin expression in duodenal biopsies was higher than normal in patients with iron overload and lower in iron-deficient patients with the exception of the inappropriately low levels found in patients with genetic hemochromatosis which is characterized by inappropriately high iron absorption [51,52]. Conversely, the cell-specific deletion of H ferritin in duodenal cells leads to unrestrained absorption and body iron overload [53], whereas ferritin overexpression caused by IRP inactivation has the opposite effect [54]. The current view is that IRP-mediated cell-autonomous regulation of ferritin synthesis sets a basal level of ferritin, which represents a temporary sink for iron not transferred to the circulation and is then lost when the apical cells are sloughed. These new findings provided a novel view of the mucosal block model proposed decades ago [55], but other control mechanisms, in particular, ferroportin-mediated basolateral transfer and the discovery of NCOA4-mediated ferritinophagy add complexity to this pathway. NCOA4 is required to avoid iron trapping in enterocytes when the demand for iron is high; however, a recent study showed quite surprisingly that in mice with intestine-specific deletions of NCOA4 iron homeostasis is not altered under normal conditions or in iron deficiency. In these settings, NCOA4 may be regulated by the HERC2 E3 ubiquitin-protein ligase, which triggers its proteasomal degradation [56]. Conversely, in a mouse model of genetic iron overload, the silencing of NCOA4 in enterocytes favored iron retention in the duodenum and mitigated systemic iron loading [57]. These findings, which are in line with the inappropriately low expression of both H and L ferritin subunits previously detected in the duodenal biopsies of patients with genetic hemochromatosis [52], suggest that the local inhibition of NCOA4 activity with consequent iron trapping within enterocytic ferritin may represent a novel therapeutic approach to limit iron uptake in the clinical conditions characterized by iron hyperabsorption.
- (c)
- Basolateral transfer. As anticipated above, the final step of intestinal iron absorption is represented by the efflux of Fe2+ at the basolateral surface, which is accomplished through conformational changes in ferroportin [58] (Figure 2). The key role of ferroportin in dietary iron absorption was shown by the rapid insurgence of anemia in adult mice in which ferroportin was specifically deleted in intestinal cells [59]. Eventually, the combined effect of two multicopper ferroxidases, membrane-bound haephestin and circulating ceruloplasmin, facilitates iron efflux and allows for Fe3+ loading onto plasma Tf for distribution [45,60]. The characterization of their role in iron absorption and mobilization provided a molecular basis for the findings of earlier elegant studies showing that a copper-deficient diet leads to iron deficiency anemia in pigs [61]. Given that strong variations in Tf saturation do not affect iron absorption in both mouse models and patients, Tf was thought to be only a passive iron acceptor. However, a recent study showing that lamina propria macrophages, in response to inflammation and iron, can produce proteases that degrade Tf locally in the interstitium, thus impairing ferroportin-dependent iron export [62], suggests that Tf may play an unexpected role in body iron absorption.
5. Regulation of Iron Absorption
6. Examples of Diseases Related to Iron Absorption
7. Therapy
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wood, B.J.; Halliday, A.N. Cooling of the Earth and core formation after the giant impact. Nature 2005, 437, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Wade, J.; Byrne, D.J.; Ballentine, C.J.; Drakesmith, H. Temporal variation of planetary iron as a driver of evolution. Proc. Natl. Acad. Sci. USA 2021, 118, e2109865118. [Google Scholar] [CrossRef] [PubMed]
- Golonka, R.; Yeoh, B.S.; Vijay-Kumar, M. The Iron Tug-of-War between Bacterial Siderophores and Innate Immunity. J. Innate. Immun. 2019, 11, 249–262. [Google Scholar] [CrossRef]
- Frank, K.M.; Schneewind, O.; Shieh, W.J. Investigation of a researcher’s death due to septicemic plague. N. Engl. J. Med. 2011, 364, 2563–2564. [Google Scholar] [CrossRef] [PubMed]
- Pasricha, S.R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet 2021, 397, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Weiss, M.J. Anemia: Progress in molecular mechanisms and therapies. Nat. Med. 2015, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Putignano, V.; Rosato, A.; Banci, L. The human iron-proteome. Metallomics 2018, 10, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Wyart, E.; Hsu, M.Y.; Sartori, R.; Mina, E.; Rausch, V.; Pierobon, E.S.; Mezzanotte, M.; Pezzini, C.; Bindels, L.B.; Lauria, A.; et al. Iron supplementation is sufficient to rescue skeletal muscle mass and function in cancer cachexia. EMBO Rep. 2022, 23, e53746. [Google Scholar] [CrossRef]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef]
- Goswami, T.; Rolfs, A.; Hediger, M.A. Iron transport: Emerging roles in health and disease. Biochem. Cell Biol. 2002, 80, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Recalcati, S.; Cairo, G. Dual Role of ROS as Signal and Stress Agents: Iron Tips the Balance in favor of Toxic Effects. Oxidative Med. Cell. Longev. 2016, 2016, 8629024. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Sindikubwabo, F.; Cañeque, T.; Lafon, A.; Versini, A.; Lombard, B.; Loew, D.; Wu, T.D.; Ginestier, C.; Charafe-Jauffret, E.; et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 2020, 12, 929–938. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends. Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Hider, R.; Aviles, M.V.; Chen, Y.L.; Latunde-Dada, G.O. The Role of GSH in Intracellular Iron Trafficking. Int. J. Mol. Sci. 2021, 22, 1278. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Merlot, A.M.; Huang, M.L.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef]
- Gao, H.; Jin, Z.; Bandyopadhyay, G.; Wang, G.; Zhang, D.; Rocha, K.C.E.; Liu, X.; Zhao, H.; Kisseleva, T.; Brenner, D.A.; et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metab. 2022, 34, 1201–1213.e5. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Ryu, M.S.; Frey, A.; Patel, S. Cytosolic iron chaperones: Proteins delivering iron cofactors in the cytosol of mammalian cells. J. Biol. Chem. 2017, 292, 12764–12771. [Google Scholar] [CrossRef]
- Arosio, P.; Carmona, F.; Gozzelino, R.; Maccarinelli, F.; Poli, M. The importance of eukaryotic ferritins in iron handling and cytoprotection. Biochem. J. 2015, 472, 1–15. [Google Scholar] [CrossRef]
- Cairo, G.; Tacchini, L.; Pogliaghi, G.; Anzon, E.; Tomasi, A.; Bernelli-Zazzera, A. Induction of ferritin synthesis by oxidative stress. Transcriptional and post-transcriptional regulation by expansion of the “free” iron pool. J. Biol. Chem. 1995, 270, 700–703. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef]
- Yanatori, I.; Richardson, D.R.; Dhekne, H.S.; Toyokuni, S.; Kishi, F. CD63 is regulated by iron via the IRE-IRP system and is important for ferritin secretion by extracellular vesicles. Blood 2021, 138, 1490–1503. [Google Scholar] [CrossRef]
- Recalcati, S.; Minotti, G.; Cairo, G. Iron regulatory proteins: From molecular mechanisms to drug development. Antioxid. Redox. Signal. 2010, 13, 1593–1616. [Google Scholar] [CrossRef]
- Sanchez, M.; Galy, B.; Muckenthaler, M.U.; Hentze, M.W. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat. Struct. Mol. Biol. 2007, 14, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Cairo, G. Macrophages and Iron: A Special Relationship. Biomedicines 2021, 9, 1585. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Ganz, T. Hepcidin and Iron in Health and Disease. Annu. Rev. Med. 2023, 74, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Erythropoietic regulators of iron metabolism. Free Radic. Biol. Med. 2019, 133, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet 2014, 46, 678–684. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Sardo, U.; Perrier, P.; Cormier, K.; Sotin, M.; Personnaz, J.; Medjbeur, T.; Desquesnes, A.; Cannizzo, L.; Ruiz-Martinez, M.; Thevenin, J.; et al. The hepatokine FGL1 regulates hepcidin and iron metabolism during anemia in mice by antagonizing BMP signaling. Blood 2024, 143, 1282–1292. [Google Scholar] [CrossRef]
- Vecchi, C.; Montosi, G.; Zhang, K.; Lamberti, I.; Duncan, S.A.; Kaufman, R.J.; Pietrangelo, A. ER stress controls iron metabolism through induction of hepcidin. Science 2009, 325, 877–880. [Google Scholar] [CrossRef]
- Chung, B.; Matak, P.; McKie, A.T.; Sharp, P. Leptin increases the expression of the iron regulatory hormone hepcidin in HuH7 human hepatoma cells. J. Nutr. 2007, 137, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, S.; Li, Q.; Wu, Y.; Jia, X.; Feng, W.; Li, Z.; Shi, Y.; Hou, Q.; Ma, J.; et al. Lactate modulates iron metabolism by binding soluble adenylyl cyclase. Cell Metab. 2023, 35, 1597–1612.e6. [Google Scholar] [CrossRef] [PubMed]
- Charlebois, E.; Pantopoulos, K. Nutritional Aspects of Iron in Health and Disease. Nutrients 2023, 15, 2441. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G397–G409. [Google Scholar] [CrossRef] [PubMed]
- Fillebeen, C.; Gkouvatsos, K.; Fragoso, G.; Calvé, A.; Garcia-Santos, D.; Buffler, M.; Becker, C.; Schümann, K.; Ponka, P.; Santos, M.M.; et al. Mice are poor heme absorbers and do not require intestinal Hmox1 for dietary heme iron assimilation. Haematologica 2015, 100, e334–e337. [Google Scholar] [CrossRef] [PubMed]
- McLaren, G.D.; Nathanson, M.H.; Jacobs, A.; Trevett, D.; Thomson, W. Regulation of intestinal iron absorption and mucosal iron kinetics in hereditary hemochromatosis. J. Lab. Clin. Med. 1991, 117, 390–401. [Google Scholar] [PubMed]
- Loechl, C.U.; Datta-Mitra, A.; Fenlason, L.; Green, R.; Hackl, L.; Itzkowitz, L.; Koso-Thomas, M.; Moorthy, D.; Owino, V.O.; Pachón, H.; et al. Approaches to Address the Anemia Challenge. J. Nutr. 2023, 153 (Suppl. S1), S42–S59. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef]
- Wang, Y.; Protchenko, O.; Huber, K.D.; Shakoury-Elizeh, M.; Ghosh, M.C.; Philpott, C.C. The iron chaperone poly(rC)-binding protein 1 regulates iron efflux through intestinal ferroportin in mice. Blood 2023, 142, 1658–1671. [Google Scholar] [CrossRef]
- Pietrangelo, A.; Rocchi, E.; Casalgrandi, G.; Rigo, G.; Ferrari, A.; Perini, M.; Ventura, E.; Cairo, G. Regulation of transferrin, transferrin receptor, and ferritin genes in human duodenum. Gastroenterology 1992, 102, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A.; Casalgrandi, G.; Quaglino, D.; Gualdi, R.; Conte, D.; Milani, S.; Montosi, G.; Cesarini, L.; Ventura, E.; Cairo, G. Duodenal ferritin synthesis in genetic hemochromatosis. Gastroenterology 1995, 108, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Vanoaica, L.; Darshan, D.; Richman, L.; Schümann, K.; Kühn, L.C. Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab. 2010, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring-Appel, D.; Becker, C.; Gretz, N.; Gröne, H.J.; Schümann, K.; Hentze, M.W. Iron regulatory proteins control a mucosal block to intestinal iron absorption. Cell Rep. 2013, 3, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Granick, S. Ferritin. 9. Increase of the protein apoferritin in the gastrointestinal mucosa as a direct response to iron feeding; the function of ferritin in the regulation of iron absorption. J. Biol. Chem. 1946, 164, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Das, N.K.; Jain, C.; Sankar, A.; Schwartz, A.J.; Santana-Codina, N.; Solanki, S.; Zhang, Z.; Ma, X.; Parimi, S.; Rui, L.; et al. Modulation of the HIF2α-NCOA4 axis in enterocytes attenuates iron loading in a mouse model of hemochromatosis. Blood 2022, 139, 2547–2552. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ren, Z.; Gao, S.; Shen, J.; Wang, L.; Xu, Z.; Yu, Y.; Bachina, P.; Zhang, H.; Fan, X.; et al. Structural basis of ion transport and inhibition in ferroportin. Nat. Commun. 2020, 11, 5686. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef]
- Lee, G.R.; Nacht, S.; Lukens, J.N.; Cartwright, G.E. Iron metabolism in copper-deficient swine. J. Clin. Investig. 1968, 47, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Schöller, M.; Fritsch, S.D.; Linke, M.; Horer, S.; Träger, M.; Mazić, M.; Forisch, S.; Gonzales, K.; Kahler, J.P.; et al. Duodenal macrophages control dietary iron absorption via local degradation of transferrin. Blood 2023, 141, 2878–2890. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D.; Trenor, C.C.; Su, M.A.; Foernzler, D.; Beier, D.R.; Dietrich, W.F.; Andrews, N.C. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997, 16, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet 1999, 21, 195–199. [Google Scholar] [CrossRef]
- Shah, Y.M.; Xie, L. Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology 2014, 146, 630–642. [Google Scholar] [CrossRef]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J. Clin. Investig. 2009, 119, 1159–1166. [Google Scholar] [CrossRef]
- Ogawa, C.; Tsuchiya, K.; Maeda, K. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors and Iron Metabolism. Int. J. Mol. Sci. 2023, 24, 3037. [Google Scholar] [CrossRef]
- Zhang, D.L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.J.; Das, N.K.; Ramakrishnan, S.K.; Jain, C.; Jurkovic, M.T.; Wu, J.; Nemeth, E.; Lakhal-Littleton, S.; Colacino, J.A.; Shah, Y.M. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J. Clin. Investig. 2019, 129, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. Iron-deficiency anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Finberg, K.E.; Heeney, M.M.; Campagna, D.R.; Aydinok, Y.; Pearson, H.A.; Hartman, K.R.; Mayo, M.M.; Samuel, S.M.; Strouse, J.J.; Markianos, K.; et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet 2008, 40, 569–571. [Google Scholar] [CrossRef] [PubMed]
- De Falco, L.; Sanchez, M.; Silvestri, L.; Kannengiesser, C.; Muckenthaler, M.U.; Iolascon, A.; Gouya, L.; Camaschella, C.; Beaumont, C. Iron refractory iron deficiency anemia. Haematologica 2013, 98, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, A.; Saha, P.K.; Saha, L. The role of TMPRSS6 gene polymorphism in iron resistance iron deficiency anaemia (IRIDA): A systematic review. Ann. Hematol. 2024, 103, 1085–1102. [Google Scholar] [CrossRef]
- Montoro-Huguet, M.A.; Belloc, B.; Domínguez-Cajal, M. Small and Large Intestine (I): Malabsorption of Nutrients. Nutrients 2021, 13, 1254. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef]
- Mahadea, D.; Adamczewska, E.; Ratajczak, A.E.; Rychter, A.M.; Zawada, A.; Eder, P.; Dobrowolska, A.; Krela-Kaźmierczak, I. Iron Deficiency Anemia in Inflammatory Bowel Diseases-A Narrative Review. Nutrients 2021, 13, 4008. [Google Scholar] [CrossRef]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Primers 2018, 4, 18016. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Pathogenic Mechanisms in Thalassemia II: Iron Overload. Hematol. Oncol. Clin. N. Am. 2023, 37, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.; Garbowski, M. Consequences and management of iron overload in sickle cell disease. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Parrow, N.L.; Violet, P.C.; George, N.A.; Ali, F.; Bhanvadia, S.; Wong, R.; Tisdale, J.F.; Fitzhugh, C.; Levine, M.; Thein, S.L.; et al. Dietary iron restriction improves markers of disease severity in murine sickle cell anemia. Blood 2021, 137, 1553–1555. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kazmi, J.S.; Lee, S.; Zhang, D.; Gao, X.; Maryanovich, M.; Torres, L.; Verma, D.; Kelly, L.; Ginzburg, Y.Z.; et al. Dietary iron restriction protects against vaso-occlusion and organ damage in murine sickle cell disease. Blood 2023, 141, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, 393–408.e2. [Google Scholar] [CrossRef] [PubMed]
- Prentice, A.M.; Mendoza, Y.A.; Pereira, D.; Cerami, C.; Wegmuller, R.; Constable, A.; Spieldenner, J. Dietary strategies for improving iron status: Balancing safety and efficacy. Nutr. Rev. 2017, 75, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Puschhof, J.; Elinav, E. Human microbiome research: Growing pains and future promises. PLoS Biol. 2023, 21, e3002053. [Google Scholar] [CrossRef] [PubMed]
- Das, N.K.; Schwartz, A.J.; Barthel, G.; Inohara, N.; Liu, Q.; Sankar, A.; Hill, D.R.; Ma, X.; Lamberg, O.; Schnizlein, M.K.; et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab. 2020, 31, 115–130.e6. [Google Scholar] [CrossRef] [PubMed]
- Khoury, A.; Pagan, K.A.; Farland, M.Z. Ferric Maltol: A New Oral Iron Formulation for the Treatment of Iron Deficiency in Adults. Ann. Pharmacother. 2021, 55, 222–229. [Google Scholar] [CrossRef]
- Fabiano, A.; Brilli, E.; Mattii, L.; Testai, L.; Moscato, S.; Citi, V.; Tarantino, G.; Zambito, Y. Ex Vivo and in Vivo Study of Sucrosomial. Int. J. Mol. Sci. 2018, 19, 2722. [Google Scholar] [CrossRef]
- Grillo, A.S.; SantaMaria, A.M.; Kafina, M.D.; Cioffi, A.G.; Huston, N.C.; Han, M.; Seo, Y.A.; Yien, Y.Y.; Nardone, C.; Menon, A.V.; et al. Restored iron transport by a small molecule promotes absorption and hemoglobinization in animals. Science 2017, 356, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, E.D. Nutritional immunity. Host’s attempt to withold iron from microbial invaders. JAMA 1975, 231, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Platre, M.P.; Tsai, H.H.; Zhang, L.; Nobori, T.; Armengot, L.; Chen, Y.; He, W.; Brent, L.; Coll, N.S.; et al. Spatial IMA1 regulation restricts root iron acquisition on MAMP perception. Nature 2024, 625, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Speich, C.; Wegmüller, R.; Brittenham, G.M.; Zeder, C.; Cercamondi, C.I.; Buhl, D.; Prentice, A.M.; Zimmermann, M.B.; Moretti, D. Measurement of long-term iron absorption and loss during iron supplementation using a stable isotope of iron. Br. J. Haematol. 2021, 192, 179–189. [Google Scholar] [CrossRef]
- Finch, C. Regulators of iron balance in humans. Blood 1994, 84, 1697–1702. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Disease | Non Genetic Cause | Mutated Gene | Function of Altered Target Protein | Molecular Basis of Altered Iron Absorption | Iron Absorption |
|---|---|---|---|---|---|
| IRIDA | - | TMPRSS6 | Negative regulation of hepcidin | Hepcidin increase | Reduced ↓ |
| IDA | Diet (major cause) | - | - | Hepcidin decrease | Increased ↑ |
| ACD | Inflammation | - | - | Hepcidin increase | Reduced ↓ |
| HH | - | HFE | Modulation of hepcidin production | Inappropriate hepcidin decrease | Increased ↑ |
| Non-HFE HH | - | HAMP | Down-regulation of ferroportin | Loss of hepcidin regulation | Increased ↑ |
| TFR2 HJV | Regulation of hepcidin expression | ||||
| SLC40A1 | Iron export | Hepcidin resistance | |||
| Thalassemia | - | HBA1 HBB | Hb formation | ERFE-mediated hepcidin repression | Increased ↑ |
| SCD | - | HBB | Hb formation | ERFE-mediated hepcidin repression (to be confirmed) | Increased ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correnti, M.; Gammella, E.; Cairo, G.; Recalcati, S. Iron Absorption: Molecular and Pathophysiological Aspects. Metabolites 2024, 14, 228. https://doi.org/10.3390/metabo14040228
Correnti M, Gammella E, Cairo G, Recalcati S. Iron Absorption: Molecular and Pathophysiological Aspects. Metabolites. 2024; 14(4):228. https://doi.org/10.3390/metabo14040228
Chicago/Turabian StyleCorrenti, Margherita, Elena Gammella, Gaetano Cairo, and Stefania Recalcati. 2024. "Iron Absorption: Molecular and Pathophysiological Aspects" Metabolites 14, no. 4: 228. https://doi.org/10.3390/metabo14040228
APA StyleCorrenti, M., Gammella, E., Cairo, G., & Recalcati, S. (2024). Iron Absorption: Molecular and Pathophysiological Aspects. Metabolites, 14(4), 228. https://doi.org/10.3390/metabo14040228