

Mitochondrial–Stem Cell Connection: Providing Additional Explanations for Understanding Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Somatic Mutation Model Alone May Not Explain Tumorigenesis
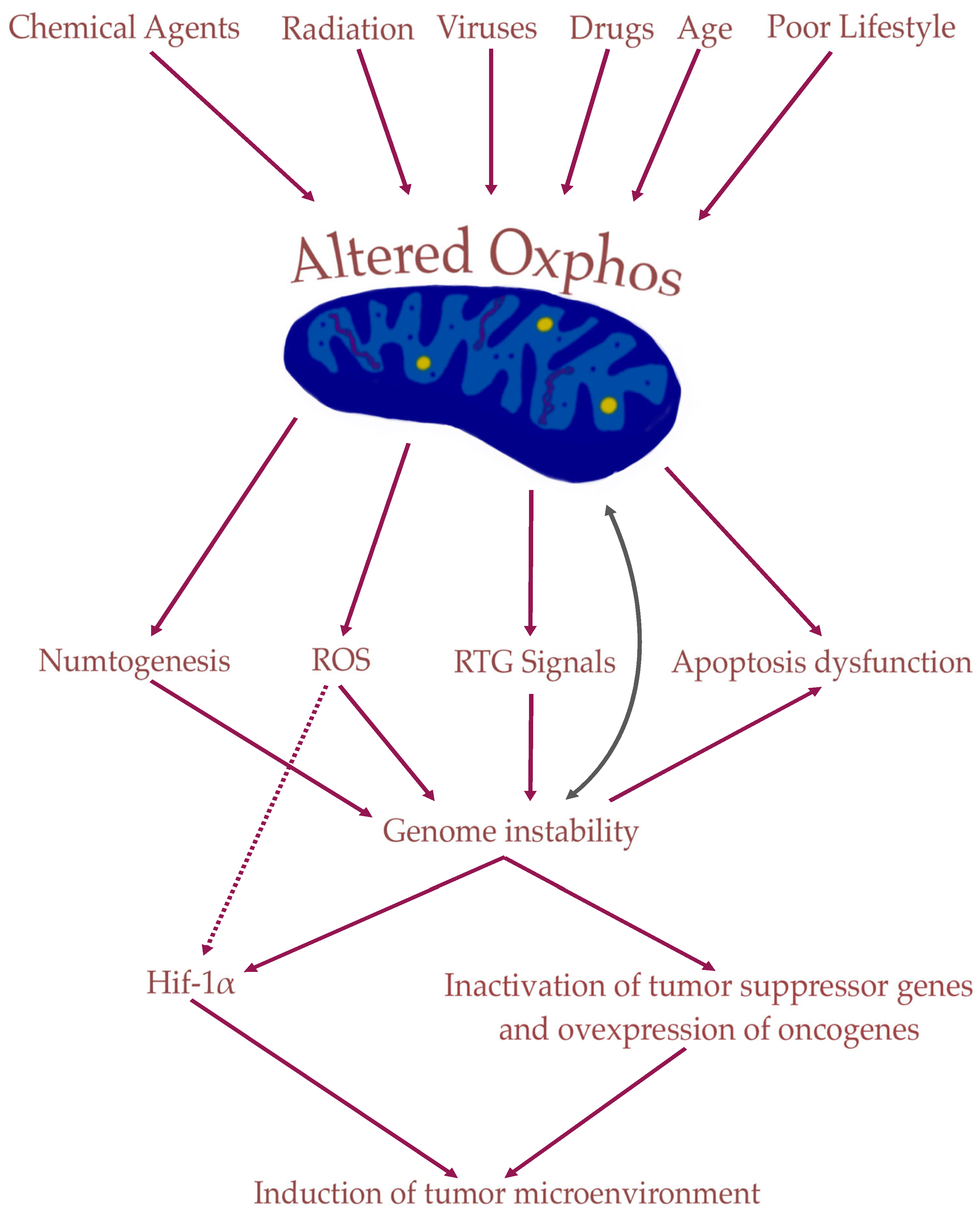
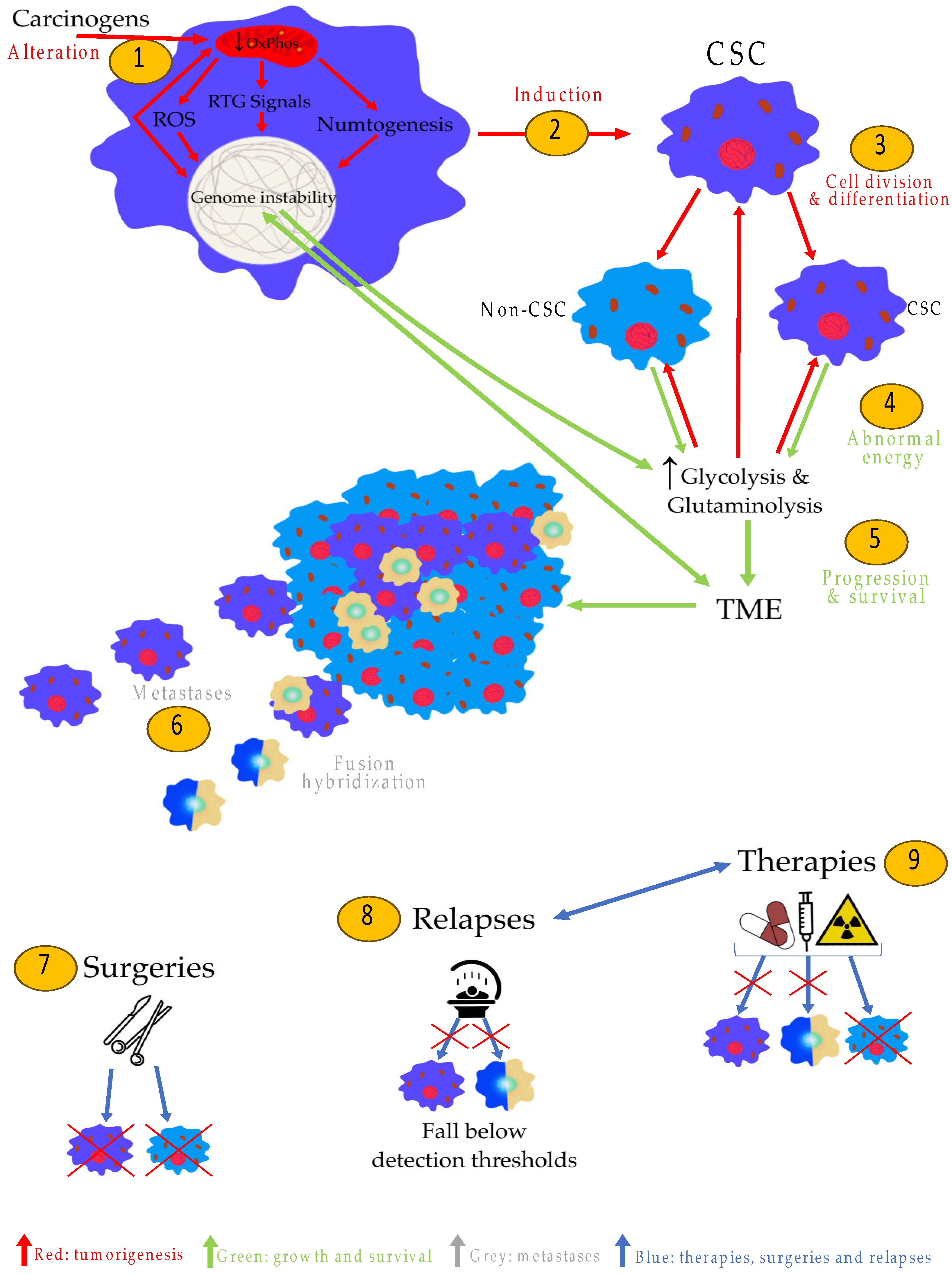
3. Role of Mitochondria in Tumorigenesis
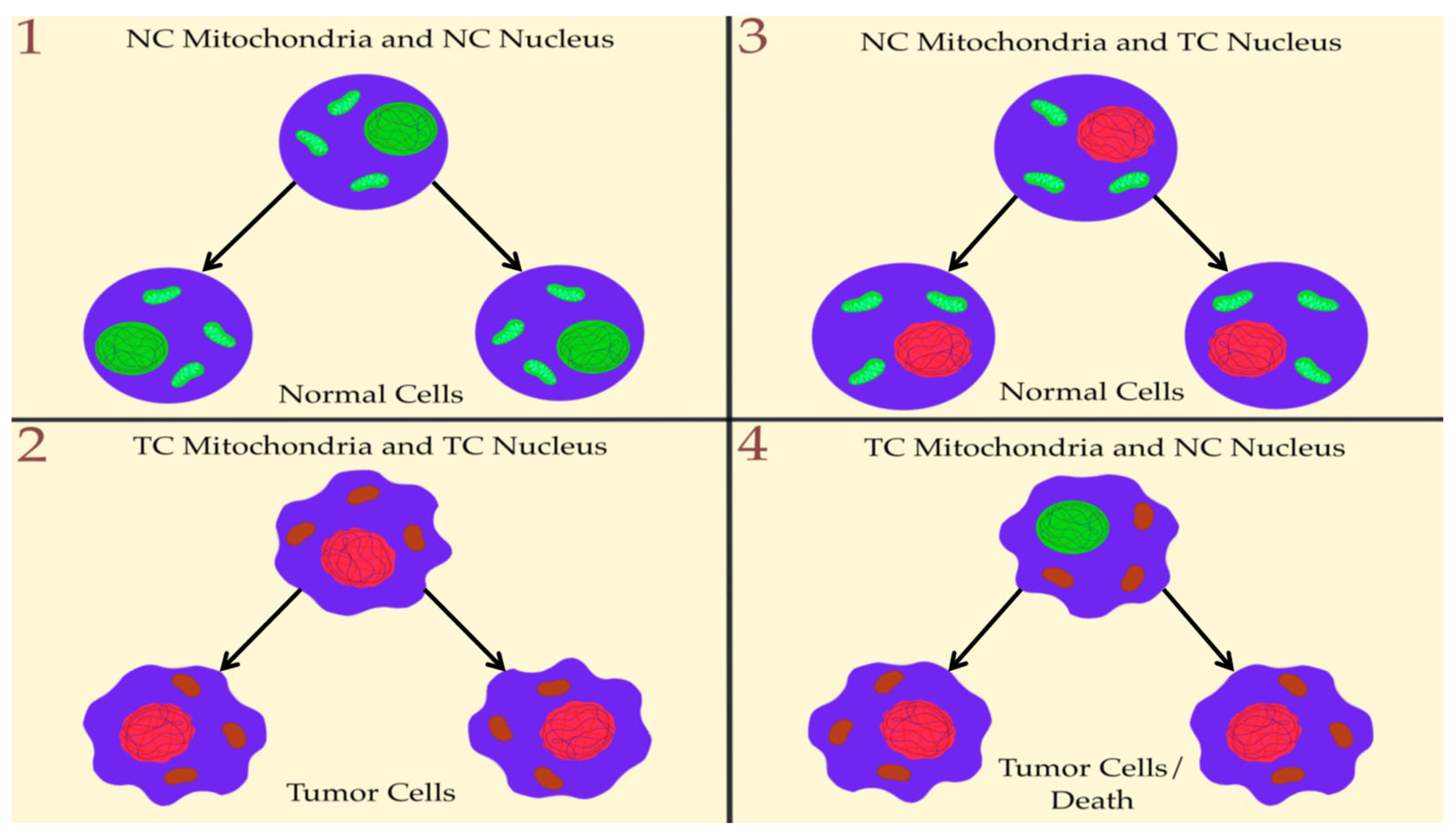
4. Nuclear Genetic Mutation Versus Mitochondrial Alteration in Tumorigenesis: Experimental Studies
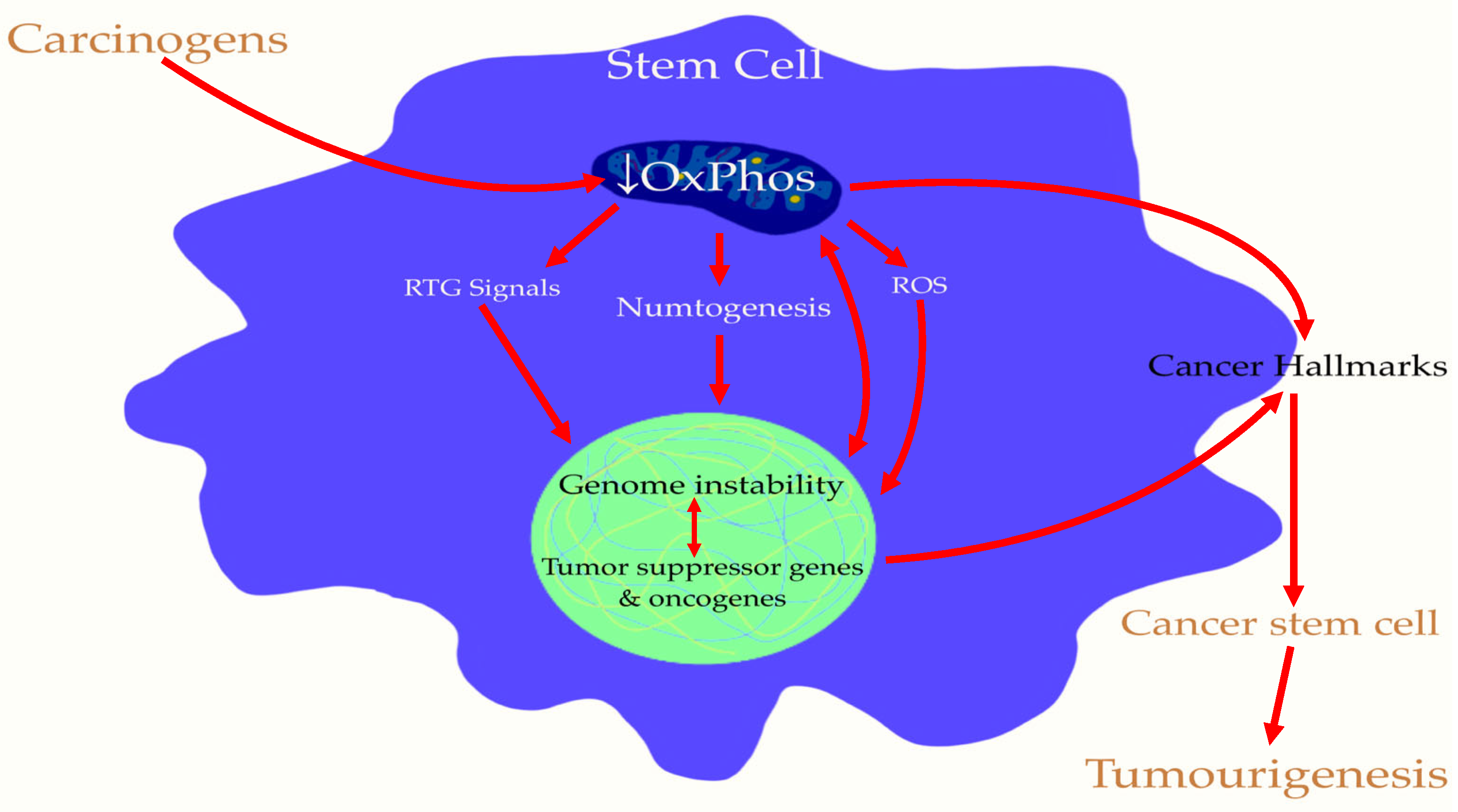
5. Cancer Stem Cells (CSCs) Are Induced by Mitochondrial Damage in Stem Cells: The Origin of Tumorigenesis
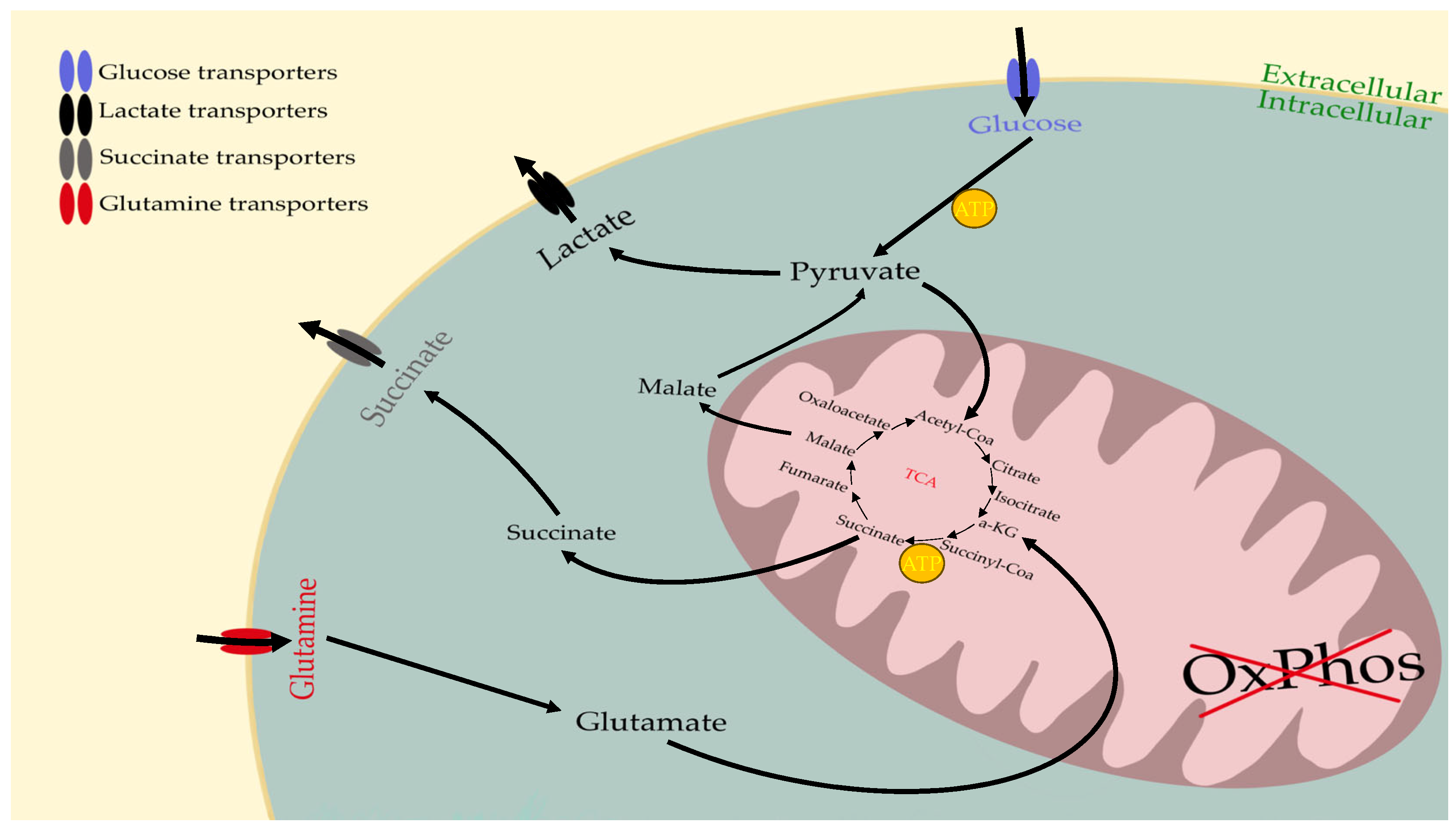
6. The Metabolism of Cancer Cells Compensates for OxPhos Insufficiency
7. CSCs and Macrophages: The Origin of Metastases
8. The Tumor Microenvironment of CSCs: A Necessary Consequence of Mitochondrial Impairment
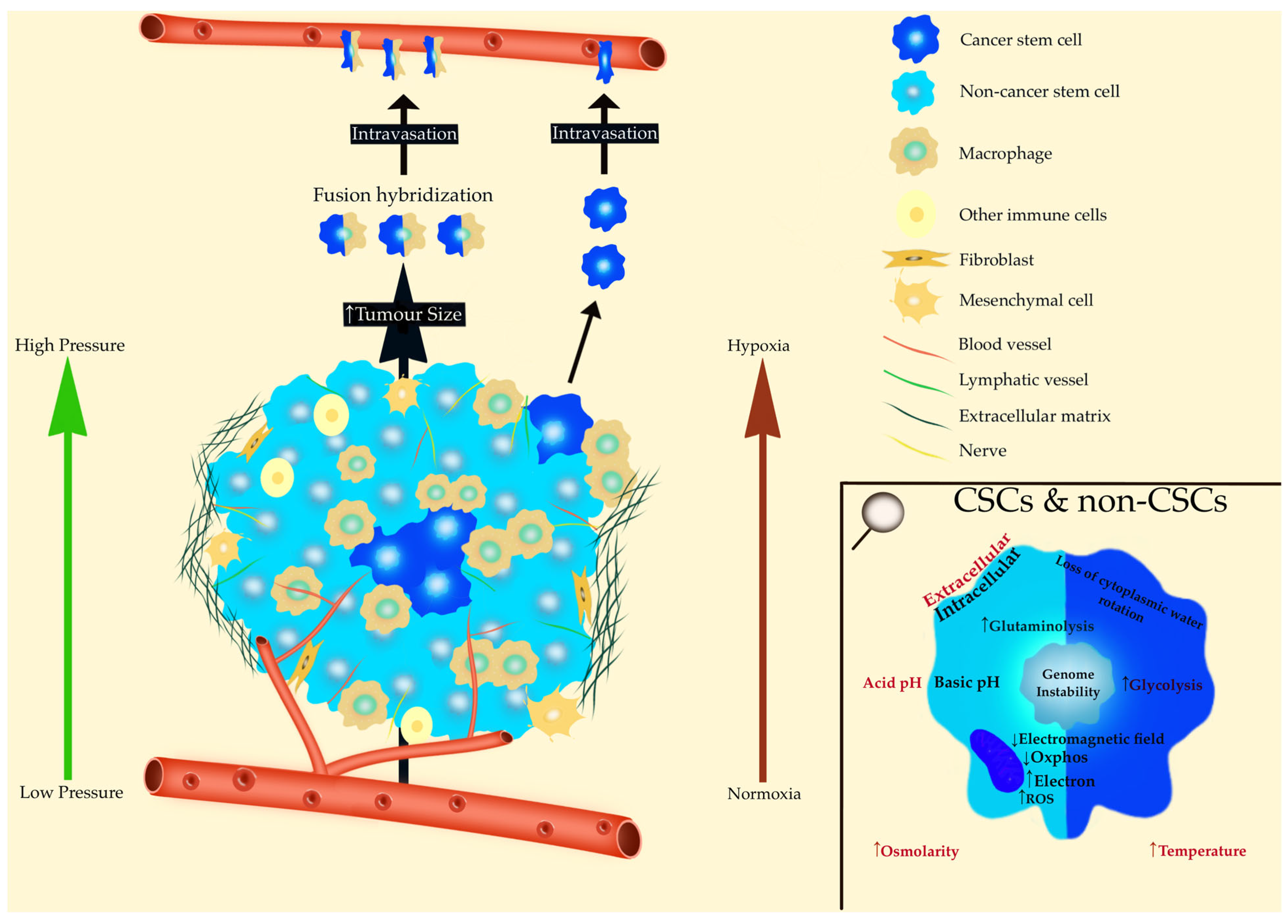
8.1. Pressure and Deformation of Tissues
8.2. pH
8.3. Hypoxia
8.4. Entropy
8.5. Temperature
8.6. Stroma
8.7. Bioelectricity and Cancer
8.8. Cytoplasmic Water
9. The CSCs (and Their Mitochondria), an Explanation of the Current Treatments and Relapses
10. Key Points of the MSCC
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Majérus, M.-A. The cause of cancer: The unifying theory. Adv. Cancer Biol.-Metastasis 2022, 4, 100034. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Chinopoulos, C. Can the Mitochondrial Metabolic Theory Explain Better the Origin and Management of Cancer than Can the Somatic Mutation Theory? Metabolites 2021, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Capp, J.P. Cancer Stem Cells: From Historical Roots to a New Perspective. J. Oncol. 2019, 2019, 5189232. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Sonnenschein, C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. Bioessays 2011, 33, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. Cancer as a global health crisis with deep evolutionary roots. Glob. Transit. 2024, 6, 45–65. [Google Scholar] [CrossRef]
- Onaciu, A.; Munteanu, R.; Munteanu, V.C.; Gulei, D.; Raduly, L.; Feder, R.I.; Pirlog, R.; Atanasov, A.G.; Korban, S.S.; Irimie, A.; et al. Spontaneous and Induced Animal Models for Cancer Research. Diagnostics 2020, 10, 660. [Google Scholar] [CrossRef]
- Zhou, Y.; Xia, J.; Xu, S.; She, T.; Zhang, Y.; Sun, Y.; Wen, M.; Jiang, T.; Xiong, Y.; Lei, J. Experimental mouse models for translational human cancer research. Front. Immunol. 2023, 14, 1095388. [Google Scholar] [CrossRef]
- Adjiri, A. DNA Mutations May Not Be the Cause of Cancer. Oncol. Ther. 2017, 5, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhang, C.; Xu, Y. Somatic mutations may not be the primary drivers of cancer formation. Int. J. Cancer 2015, 137, 2762–2765. [Google Scholar] [CrossRef] [PubMed]
- Woodman, S.E.; Mills, G.B. Are oncogenes sufficient to cause human cancer? Proc. Natl. Acad. Sci. USA 2010, 107, 20599–20600. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.; Cairns, J. The mysterious steps in carcinogenesis. Br. J. Cancer 2009, 101, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Balmain, A.; Harris, C.C. Carcinogenesis in mouse and human cells: Parallels and paradoxes. Carcinogenesis 2000, 21, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.P.; Holt, S.E.; Ouellette, M.; Kaur, K.J.; Yan, Y.; Wilson, K.S.; White, M.A.; Wright, W.E.; Shay, J.W. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat. Genet. 1999, 21, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Plattner, R.; Anderson, M.J.; Sato, K.Y.; Fasching, C.L.; Der, C.J.; Stanbridge, E.J. Loss of oncogenic ras expression does not correlate with loss of tumorigenicity in human cells. Proc. Natl. Acad. Sci. USA 1996, 93, 6665–6670. [Google Scholar] [CrossRef] [PubMed]
- de Magalhães, J.P. Every gene can (and possibly will) be associated with cancer. Trends Genet. 2022, 38, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2658 human cancer genomes. Cell 2021, 184, 2239–2254.e39. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726. [Google Scholar] [CrossRef]
- Li, C.; Williams, S.M. Human Somatic Variation: It’s Not Just for Cancer Anymore. Curr. Genet. Med. Rep. 2013, 1, 212–218. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, W.; Velculescu, V.E.; Kern, S.E.; Hruban, R.H.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Gene expression profiles in normal and cancer cells. Science 1997, 276, 1268–1272. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Chanock, S.J. The paradox of mutations and cancer. Science 2018, 362, 893–894. [Google Scholar] [CrossRef]
- Gormally, E.; Vineis, P.; Matullo, G.; Veglia, F.; Caboux, E.; Le Roux, E.; Peluso, M.; Garte, S.; Guarrera, S.; Munnia, A.; et al. TP53 and KRAS2 mutations in plasma DNA of healthy subjects and subsequent cancer occurrence: A prospective study. Cancer Res. 2006, 66, 6871–6876. [Google Scholar] [CrossRef] [PubMed]
- Krimmel, J.D.; Schmitt, M.W.; Harrell, M.I.; Agnew, K.J.; Kennedy, S.R.; Emond, M.J.; Loeb, L.A.; Swisher, E.M.; Risques, R.A. Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 6005–6010. [Google Scholar] [CrossRef] [PubMed]
- Rème, T.; Travaglio, A.; Gueydon, E.; Adla, L.; Jorgensen, C.; Sany, J. Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin. Exp. Immunol. 1998, 111, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Boyle, D.L.; Rosengren, S.; Green, D.R.; Zvaifler, N.J.; Firestein, G.S. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 2002, 99, 10025–10030. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; Echeverri, F.; Yeo, M.; Zvaifler, N.J.; Green, D.R. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900. [Google Scholar] [CrossRef] [PubMed]
- Mendiratta, G.; Ke, E.; Aziz, M.; Liarakos, D.; Tong, M.; Stites, E.C. Cancer gene mutation frequencies for the U.S. population. Nat. Commun. 2021, 12, 5961. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Sonnenschein, C. The somatic mutation theory of cancer: Growing problems with the paradigm? Bioessays 2004, 26, 1097–1107. [Google Scholar] [CrossRef]
- Hernández, L.G.; van Steeg, H.; Luijten, M.; van Benthem, J. Mechanisms of non-genotoxic carcinogens and importance of a weight of evidence approach. Mutat. Res. 2009, 682, 94–109. [Google Scholar] [CrossRef]
- Lijinsky, W. Non-genotoxic environmental carcinogens. Environ. Carcinog. Rev. 1990, 8, 45–87. [Google Scholar] [CrossRef]
- Bischoff, F.; Bryson, G. Carcinogenesis through solid state surfaces. Prog. Exp. Tumor. Res. 1964, 5, 85–133. [Google Scholar] [CrossRef]
- Mally, A.; Chipman, J.K. Non-genotoxic carcinogens: Early effects on gap junctions, cell proliferation and apoptosis in the rat. Toxicology 2002, 180, 233–248. [Google Scholar] [CrossRef]
- Monti, N.; Verna, R.; Piombarolo, A.; Querqui, A.; Bizzarri, M.; Fedeli, V. Paradoxical Behavior of Oncogenes Undermines the Somatic Mutation Theory. Biomolecules 2022, 12, 662. [Google Scholar] [CrossRef]
- Brücher, B.L.D.M.; Jamall, I.S. Somatic Mutation Theory—Why it’s Wrong for Most Cancers. Cell. Physiol. Biochem. 2016, 38, 1663–1680. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef]
- Kuretu, A.; Arineitwe, C.; Mothibe, M.; Ngubane, P.; Khathi, A.; Sibiya, N. Drug-induced mitochondrial toxicity: Risks of developing glucose handling impairments. Front. Endocrinol. 2023, 14, 1123928. [Google Scholar] [CrossRef]
- Hoogstraten, C.A.; Lyon, J.J.; Smeitink, J.A.M.; Russel, F.G.M.; Schirris, T.J.J. Time to Change: A Systems Pharmacology Approach to Disentangle Mechanisms of Drug-Induced Mitochondrial Toxicity. Pharmacol. Rev. 2023, 75, 463–486. [Google Scholar] [CrossRef] [PubMed]
- Reddam, A.; McLarnan, S.; Kupsco, A. Environmental Chemical Exposures and Mitochondrial Dysfunction: A Review of Recent Literature. Curr. Environ. Health Rep. 2022, 9, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Kam, W.W.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Elesela, S.; Lukacs, N.W. Role of Mitochondria in Viral Infections. Life 2021, 11, 232. [Google Scholar] [CrossRef]
- El Sayed, S.M. Biochemical Origin of the Warburg Effect in Light of 15 Years of Research Experience: A Novel Evidence-Based View (An Expert Opinion Article). OncoTargets Ther. 2023, 16, 143–155. [Google Scholar] [CrossRef]
- Iranmanesh, Y.; Jiang, B.; Favour, O.C.; Dou, Z.; Wu, J.; Li, J.; Sun, C. Mitochondria’s Role in the Maintenance of Cancer Stem Cells in Glioblastoma. Front. Oncol. 2021, 11, 582694. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Kulawiec, M.; Singh, K.K. Mitochondria and human cancer. Curr. Mol. Med. 2007, 7, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Mahmood, M.; Reznik, E.; Gammage, P.A. Mitochondrial DNA is a major source of driver mutations in cancer. Trends Cancer 2022, 8, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Seyfried, T.N. Absence of pathogenic mitochondrial DNA mutations in mouse brain tumors. BMC Cancer 2005, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Bermúdez, A.; Vallejo, C.G.; Vicente-Blanco, R.J.; Gallardo, M.E.; Fernández-Moreno, M.; Quintanilla, M.; Garesse, R. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget 2015, 6, 13628–13643. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L. Tumor mitochondria and the bioenergetics of cancer cells. Prog. Exp. Tumor. Res. 1978, 22, 190–274. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria organelle transplantation: Introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in human cancer. Int. J. Biochem. Cell Biol. 2009, 41, 2062–2068. [Google Scholar] [CrossRef]
- Deighton, R.F.; Le Bihan, T.; Martin, S.F.; Gerth, A.M.J.; McCulloch, M.; Edgar, J.M.; Kerr, L.E.; Whittle, I.R.; McCulloch, J. Interactions among mitochondrial proteins altered in glioblastoma. J. Neurooncol. 2014, 118, 247–256. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Arismendi-Morillo, G.; Mukherjee, P.; Chinopoulos, C. On the Origin of ATP Synthesis in Cancer. iScience 2020, 23, 101761. [Google Scholar] [CrossRef] [PubMed]
- Badrinath, N.; Yoo, S.Y. Mitochondria in cancer: In the aspects of tumorigenesis and targeted therapy. Carcinogenesis 2018, 39, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Buss, L.G.; Rheinheimer, B.A.; Limesand, K.H. Radiation-induced changes in energy metabolism result in mitochondrial dysfunction in salivary glands. Sci. Rep. 2024, 14, 845. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.J.; Marsman, D.S.; Popp, J.A.; Thurman, R.G. Several nongenotoxic carcinogens uncouple mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta (BBA)—Bioenerg. 1992, 1102, 237–244. [Google Scholar] [CrossRef]
- Ito, Y.; Nakajima, K.; Masubuchi, Y.; Kikuchi, S.; Saito, F.; Akahori, Y.; Jin, M.; Yoshida, T.; Shibutani, M. Differential responses on energy metabolic pathway reprogramming between genotoxic and non-genotoxic hepatocarcinogens in rat liver cells. J. Toxicol. Pathol. 2019, 32, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Roskelley, R.C.; Mayer, N.; Horwitt, B.N.; Salter, W.T. Studies in cancer. VII. Enzyme deficiency in human and experimental cancer. J. Clin. Investig. 1943, 22, 743–751. [Google Scholar] [CrossRef]
- Jazwinski, S.M. The retrograde response: A conserved compensatory reaction to damage from within and from without. Prog. Mol. Biol. Transl. Sci. 2014, 127, 133–154. [Google Scholar] [CrossRef]
- da Cunha, F.M.; Torelli, N.Q.; Kowaltowski, A.J. Mitochondrial Retrograde Signaling: Triggers, Pathways, and Outcomes. Oxid. Med. Cell. Longev. 2015, 2015, 482582. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kim, J. Mitochondrial Retrograde Signalling and Metabolic Alterations in the Tumour Microenvironment. Cells 2019, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Choudhury, A.R.; Tiwari, H.K. Numtogenesis as a mechanism for development of cancer. Semin. Cancer Biol. 2017, 47, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Puertas, M.J.; González-Sánchez, M. Insertions of mitochondrial DNA into the nucleus—Effects and role in cell evolution. Genome 2020, 63, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Missiroli, S.; Perrone, M.; Fiorica, F.; Pinton, P.; Giorgi, C. Mitochondrial Control of Genomic Instability in Cancer. Cancers 2021, 13, 1914. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Graziano, V.; Galavotti, S.; Henriquez, N.V.; Betts, J.; Saxena, J.; Minieri, V.; Deli, A.; Karlsson, A.; Martins, L.M.; et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Singh, K.K. Genetic insights into OXPHOS defect and its role in cancer. Biochim. Biophys. Acta (BBA)—Bioenerg. 2011, 1807, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Koura, M.; Isaka, H.; Yoshida, M.C.; Tosu, M.; Sekiguchi, T. Suppression of tumorigenicity in interspecific reconstituted cells and cybrids. Gan 1982, 73, 574–580. [Google Scholar] [PubMed]
- Israel, B.A.; Schaeffer, W.I. Cytoplasmic suppression of malignancy. Vitro Cell. Dev. Biol. 1987, 23, 627–632. [Google Scholar] [CrossRef]
- Shay, J.W.; Werbin, H. Cytoplasmic suppression of tumorigenicity in reconstructed mouse cells. Cancer Res. 1988, 48, 830–833. [Google Scholar]
- Howell, A.N.; Sager, R. Tumorigenicity and its suppression in cybrids of mouse and Chinese hamster cell lines. Proc. Natl. Acad. Sci. USA 1978, 75, 2358–2362. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, J.; Harris, H. The analysis of malignancy by cell fusion. VIII. Evidence for the intervention of an extra-chromosomal element. J. Cell Sci. 1977, 24, 255–263. [Google Scholar] [CrossRef] [PubMed]
- McKinnell, R.G.; Deggins, B.A.; Labat, D.D. Transplantation of pluripotential nuclei from triploid frog tumors. Science 1969, 165, 394–396. [Google Scholar] [CrossRef] [PubMed]
- Mintz, B.; Illmensee, K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. USA 1975, 72, 3585–3589. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Connelly, M.C.; Wetmore, C.; Curran, T.; Morgan, J.I. Mouse embryos cloned from brain tumors. Cancer Res. 2003, 63, 2733–2736. [Google Scholar] [PubMed]
- Hochedlinger, K.; Blelloch, R.; Brennan, C.; Yamada, Y.; Kim, M.; Chin, L.; Jaenisch, R. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004, 18, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Maffini, M.V.; Calabro, J.M.; Soto, A.M.; Sonnenschein, C. Stromal regulation of neoplastic development: Age-dependent normalization of neoplastic mammary cells by mammary stroma. Am. J. Pathol. 2005, 167, 1405–1410. [Google Scholar] [CrossRef]
- Coleman, W.B.; Wennerberg, A.E.; Smith, G.J.; Grisham, J.W. Regulation of the differentiation of diploid and some aneuploid rat liver epithelial (stemlike) cells by the hepatic microenvironment. Am. J. Pathol. 1993, 142, 1373–1382. [Google Scholar]
- McCullough, K.D.; Coleman, W.B.; Smith, G.J.; Grisham, J.W. Age-dependent induction of hepatic tumor regression by the tissue microenvironment after transplantation of neoplastically transformed rat liver epithelial cells into the liver. Cancer Res. 1997, 57, 1807–1813. [Google Scholar]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.Y.; Wong, L.J. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS ONE 2013, 8, e61747. [Google Scholar] [CrossRef]
- Israel, B.A.; Schaeffer, W.I. Cytoplasmic mediation of malignancy. Vitro Cell. Dev. Biol. 1988, 24, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Darlington, C.D. The plasmagene theory of the origin of cancer. Br. J. Cancer 1948, 2, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Chang, H.S.; Wu, Y.C.; Cheng, W.L.; Lin, T.T.; Chang, H.J.; Kuo, S.J.; Chen, S.T.; Liu, C.S. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 30. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Hou, Y.; Yu, Z.; Zhao, Z.; Liu, Z. Healthy mitochondria inhibit the metastatic melanoma in lungs. Int. J. Biol. Sci. 2019, 15, 2707–2718. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Bai, R.K.; Trieu, R.; Wong, L.J. Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochim. Biophys. Acta (BBA)—Bioenerg. 2010, 1797, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019, 9, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Oxidative metabolism in cancer growth. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Cuezva, J.M.; Ortega, A.D.; Willers, I.; Sánchez-Cenizo, L.; Aldea, M.; Sánchez-Aragó, M. The tumor suppressor function of mitochondria: Translation into the clinics. Biochim. Biophys. Acta (BBA)—Bioenerg. 2009, 1792, 1145–1158. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Amador-Martinez, I.; Maycotte, P. Mitochondrial transplantation strategies in multifaceted induction of cancer cell death. Life Sci. 2023, 332, 122098. [Google Scholar] [CrossRef]
- Ginestier, C. Les cellules souches cancéreuse: Définition et techniques d’isolement. Bull. Cancer. 2017, 104, 1060–1063. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Strasser, A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. 2008, 68, 4018–4021. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Aoyagi, M.; Ando, N.; Ogishima, T.; Wakimoto, H.; Yamamoto, M.; Ohno, K. Expansion of CD133-positive glioma cells in recurrent de novo glioblastomas after radiotherapy and chemotherapy. J. Neurosurg. 2013, 119, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Huels, D.J.; Sansom, O.J. Stem vs non-stem cell origin of colorectal cancer. Br. J. Cancer 2015, 113, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.C.; Noronha, S.M. Cancer stem cells: A new approach to tumor development. Rev. Assoc. Med. Bras. 2015, 61, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Garimella, S.V.; Gampa, S.C.; Chaturvedi, P. Mitochondria in Cancer Stem Cells: From an Innocent Bystander to a Central Player in Therapy Resistance. Stem Cells Cloning 2023, 16, 19–41. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Xu, D.; Tiek, D.; Goenka, A.; Wu, B.; Sastry, N.; Hu, B.; Cheng, S.Y. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020, 10, 8721–8743. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Toledo-Guzmán, M.E.; Bigoni-Ordóñez, G.D.; Ibáñez Hernández, M.; Ortiz-Sánchez, E. Cancer stem cell impact on clinical oncology. World J. Stem Cells 2018, 10, 183–195. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Hurt, E.M.; Kawasaki, B.T.; Klarmann, G.J.; Thomas, S.B.; Farrar, W.L. CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br. J. Cancer 2008, 98, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Jagust, P.; de Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, P.P.; Patro, B.S.; Bhutia, S.K. Dysregulation of mitophagy and mitochondrial homeostasis in cancer stem cells: Novel mechanism for anti-cancer stem cell-targeted cancer therapy. Br. J. Pharmacol. 2022, 179, 5015–5035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef]
- Adams, P.D.; Jasper, H.; Rudolph, K.L. Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Cell Stem Cell 2015, 16, 601–612. [Google Scholar] [CrossRef]
- Sell, S. On the stem cell origin of cancer. Am. J. Pathol. 2010, 176, 2584–2594. [Google Scholar] [CrossRef]
- Fan, M.; Shi, Y.; Zhao, J.; Li, L. Cancer stem cell fate determination: Mito-nuclear communication. Cell Commun. Signal. 2023, 21, 159. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [PubMed]
- Shevah, O.; Laron, Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: A preliminary report. Growth Horm. IGF Res. 2007, 17, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Guevara, C.; Guevara, A.; Gavilanes, A.A. Branding of subjects affected with genetic syndromes of severe short stature in developing countries. BMJ Case Rep. 2020, 13, e231737. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 2019, 54, 100940. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Shin, D.M.; Wan, W.; Liu, R.; Masternak, M.M.; Piotrowska, K.; Wiszniewska, B.; Kucia, M.; Bartke, A.; Ratajczak, M.Z. Higher number of stem cells in the bone marrow of circulating low Igf-1 level Laron dwarf mice--novel view on Igf-1, stem cells and aging. Leukemia 2011, 25, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Brown-Borg, H.M.; Johnson, W.T.; Rakoczy, S.G. Expression of oxidative phosphorylation components in mitochondria of long-living Ames dwarf mice. Age 2012, 34, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Kuo, Y.C.; Chuong, C.M.; Huang, Y.H. Niche Modulation of IGF-1R Signaling: Its Role in Stem Cell Pluripotency, Cancer Reprogramming, and Therapeutic Applications. Front. Cell Dev. Biol. 2020, 8, 625943. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Y.; Guo, Y.; Shi, X.; Chen, X.; Feng, W.; Wu, L.L.; Zhang, J.; Yu, S.; Wang, Y.; et al. An Overview: The Diversified Role of Mitochondria in Cancer Metabolism. Int. J. Biol. Sci. 2023, 19, 897–915. [Google Scholar] [CrossRef]
- Chae, H.S.; Hong, S.T. Overview of Cancer Metabolism and Signaling Transduction. Int. J. Mol. Sci. 2022, 24, 12. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Inguva, A.; Jordan, C.T. Targeting Energy Metabolism in Cancer Stem Cells: Progress and Challenges in Leukemia and Solid Tumors. Cell Stem Cell 2021, 28, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Garde, A.; Sherwood, D.R. Fueling Cell Invasion through Extracellular Matrix. Trends. Cell Biol. 2021, 31, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Busk, M.; Horsman, M.R.; Kristjansen, P.E.; van der Kogel, A.J.; Bussink, J.; Overgaard, J. Aerobic glycolysis in cancers: Implications for the usability of oxygen-responsive genes and fluorodeoxyglucose-PET as markers of tissue hypoxia. Int. J. Cancer 2008, 122, 2726–2734. [Google Scholar] [CrossRef] [PubMed]
- González, M.J.; Rosario-Pérez, G.; Guzmán, A.M.; Miranda-Massari, J.R.; Duconge, J.; Lavergne, J.; Fernandez, N.; Ortiz, N.; Quintero, A.; Mikirova, N.; et al. Mitochondria, Energy and Cancer: The Relationship with Ascorbic Acid. J. Orthomol. Med. 2010, 25, 29–38. [Google Scholar]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Peña, G.; Pazmiño, G.; Acosta, W.; Saavedra, J.; Lescano, D.; Guevara, A.; Gavilanes, A.W.D. Cancer in Ecuadorian subjects with Laron syndrome (ELS). Endocr. Relat. Cancer 2023, 30, e220389. [Google Scholar] [CrossRef]
- Lukey, M.J.; Wilson, K.F.; Cerione, R.A. Therapeutic strategies impacting cancer cell glutamine metabolism. Future Med. Chem. 2013, 5, 1685–1700. [Google Scholar] [CrossRef]
- Liao, J.; Liu, P.-P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through β-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, H.S.; Somasagara, R.R.; Bunting, K.D. Targeting MYC Dependence by Metabolic Inhibitors in Cancer. Genes 2017, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Griffeth, L.K. Use of PET/CT scanning in cancer patients: Technical and practical considerations. Proc. (Bayl. Univ. Med. Cent.) 2005, 18, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ploessl, K.; Zhou, R.; Mankoff, D.; Kung, H.F. Metabolic Imaging of Glutamine in Cancer. J. Nucl. Med. 2017, 58, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C. From Glucose to Lactate and Transiting Intermediates Through Mitochondria, Bypassing Pyruvate Kinase: Considerations for Cells Exhibiting Dimeric PKM2 or Otherwise Inhibited Kinase Activity. Front. Physiol. 2020, 11, 543564. [Google Scholar] [CrossRef] [PubMed]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef] [PubMed]
- Abd, G.M.; Laird, M.C.; Ku, J.C.; Li, Y. Hypoxia-induced cancer cell reprogramming: A review on how cancer stem cells arise. Front. Oncol. 2023, 13, 1227884. [Google Scholar] [CrossRef]
- Qian, J.; Rankin, E.B. Hypoxia-Induced Phenotypes that Mediate Tumor Heterogeneity. Adv. Exp. Med. Biol. 2019, 1136, 43–55. [Google Scholar] [CrossRef]
- Duraj, T.; Carrión-Navarro, J.; Seyfried, T.N.; García-Romero, N.; Ayuso-Sacido, A. Metabolic therapy and bioenergetic analysis: The missing piece of the puzzle. Mol. Metab. 2021, 54, 101389. [Google Scholar] [CrossRef] [PubMed]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K.; et al. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab. 2019, 29, 399–416.e10. [Google Scholar] [CrossRef] [PubMed]
- Ta, N.L.; Seyfried, T.N. Influence of Serum and Hypoxia on Incorporation of [(14)C]-D-Glucose or [(14)C]-L-Glutamine into Lipids and Lactate in Murine Glioblastoma Cells. Lipids 2015, 50, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Renner, C.; Asperger, A.; Seyffarth, A.; Meixensberger, J.; Gebhardt, R.; Gaunitz, F. Carnosine inhibits ATP production in cells from malignant glioma. Neurol. Res. 2010, 32, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Barron, E.S. The catalytic effect of methylene blue on the oxygen consumption of tumors and normal tissues. J. Exp. Med. 1930, 52, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Ceruti, S.; Mazzola, A.; Abbracchio, M.P. Resistance of human astrocytoma cells to apoptosis induced by mitochondria-damaging agents: Possible implications for anticancer therapy. J. Pharmacol. Exp. Ther. 2005, 314, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Mathews, E.H.; Stander, B.A.; Joubert, A.M.; Liebenberg, L. Tumor cell culture survival following glucose and glutamine deprivation at typical physiological concentrations. Nutrition 2014, 30, 218–227. [Google Scholar] [CrossRef]
- Holm, E.; Hagmüller, E.; Staedt, U.; Schlickeiser, G.; Günther, H.; Leweling, H.; Tokus, M.; Kollmar, H. Substrate balances across colonic carcinomas in Humans. Cancer Res. 1995, 55, 1373–1378. [Google Scholar] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Ruiz, J.; Xing, F.; Lo, H.W.; Craddock, L.; Pullikuth, A.K.; Miller, L.D.; Soike, M.H.; O’Neill, S.S.; Watabe, K.; et al. Single-cell sequencing reveals the landscape of the human brain metastatic microenvironment. Commun. Biol. 2023, 6, 760. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, X.; Huang, X.; Zhang, D.; Chen, Z.; Zhang, J.; Bai, R.; Zhang, S.; Zhao, H.; Xu, Z.; et al. Single-cell transcriptomic analysis deciphers heterogenous cancer stem-like cells in colorectal cancer and their organ-specific metastasis. Gut 2024, 73, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.W.; Zhang, H.; Xu, D.; Chen, J.X.; Chen, W.J.; Gan, S.S.; Qu, F.J.; Chu, C.M.; Cao, J.W.; Fan, Y.H.; et al. Identification of a novel cancer stem cell subpopulation that promotes progression of human fatal renal cell carcinoma by single-cell RNA-seq analysis. Int. J. Biol. Sci. 2020, 16, 3149–3162. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Verga Falzacappa, M.V.; Ronchini, C.; Reavie, L.B.; Pelicci, P.G. Regulation of self-renewal in normal and cancer stem cells. FEBS J. 2012, 279, 3559–3572. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Savic, D.; Dudás, J.; Kvitsaridze, I.; Skvortsov, S.; Riechelmann, H.; Skvortsova, I.I. Cancer stem cells and their unique role in metastatic spread. Semin Cancer Biol. 2020, 60, 148–156. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, L.A.; Montalbán-Hernández, K.; Avendaño-Ortiz, J.; Marín, E.; Lozano, R.; Toledano, V.; Sánchez-Maroto, L.; Terrón, V.; Valentín, J.; Pulido, E.; et al. Tumor stem cells fuse with monocytes to form highly invasive tumor-hybrid cells. Oncoimmunology 2020, 9, 1773204. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Aramini, B.; Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Morandi, U.; Dominici, M.; Haider, K.H. Cancer stem cells and macrophages: Molecular connections and future perspectives against cancer. Oncotarget 2021, 12, 230–250. [Google Scholar] [CrossRef]
- Allavena, P.; Digifico, E.; Belgiovine, C. Macrophages and cancer stem cells: A malevolent alliance. Mol. Med. 2021, 27, 121. [Google Scholar] [CrossRef]
- Sharma, V.P.; Tang, B.; Wang, Y.; Duran, C.L.; Karagiannis, G.S.; Xue, E.A.; Entenberg, D.; Borriello, L.; Coste, A.; Eddy, R.J.; et al. Live tumor imaging shows macrophage induction and TMEM-mediated enrichment of cancer stem cells during metastatic dissemination. Nat. Commun. 2021, 12, 7300. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Alibert, C.; Goud, B.; Manneville, J.B. Are cancer cells really softer than normal cells? Biol. Cell 2017, 109, 167–189. [Google Scholar] [CrossRef]
- Burgdorf, S.; Porubsky, S.; Marx, A.; Popovic, Z.V. Cancer Acidity and Hypertonicity Contribute to Dysfunction of Tumor-Associated Dendritic Cells: Potential Impact on Antigen Cross-Presentation Machinery. Cancers 2020, 12, 2403. [Google Scholar] [CrossRef]
- Follain, G.; Herrmann, D.; Harlepp, S.; Hyenne, V.; Osmani, N.; Warren, S.C.; Timpson, P.; Goetz, J.G. Fluids and their mechanics in tumour transit: Shaping metastasis. Nat. Rev. Cancer 2020, 20, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Frieboes, H.B.; McDougall, S.R.; Chaplain, M.A.; Cristini, V.; Lowengrub, J. The effect of interstitial pressure on tumor growth: Coupling with the blood and lymphatic vascular systems. J. Theor. Biol. 2013, 320, 131–151. [Google Scholar] [CrossRef]
- Devic, S. Warburg Effect—A Consequence or the Cause of Carcinogenesis? J. Cancer 2016, 7, 817–822. [Google Scholar] [CrossRef]
- Morita, M.; Sato, T.; Nomura, M.; Sakamoto, Y.; Inoue, Y.; Tanaka, R.; Ito, S.; Kurosawa, K.; Yamaguchi, K.; Sugiura, Y.; et al. PKM1 Confers Metabolic Advantages and Promotes Cell-Autonomous Tumor Cell Growth. Cancer Cell 2018, 33, 355–367.e7. [Google Scholar] [CrossRef] [PubMed]
- Marchiq, I.; Pouysségur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. 2016, 94, 155–171. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Arismendi-Morillo, G.; Zuccoli, G.; Lee, D.C.; Duraj, T.; Elsakka, A.M.; Maroon, J.C.; Mukherjee, P.; Ta, L.; Shelton, L.; et al. Metabolic management of microenvironment acidity in glioblastoma. Front. Oncol. 2022, 12, 968351. [Google Scholar] [CrossRef]
- Farhana, A.; Alsrhani, A.; Khan, Y.S.; Rasheed, Z. Cancer Bioenergetics and Tumor Microenvironments—Enhancing Chemotherapeutics and Targeting Resistant Niches through Nanosystems. Cancers 2023, 15, 3836. [Google Scholar] [CrossRef] [PubMed]
- da Veiga Moreira, J.; De Staercke, L.; César Martínez-Basilio, P.; Gauthier-Thibodeau, S.; Montégut, L.; Schwartz, L.; Jolicoeur, M. Hyperosmolarity Triggers the Warburg Effect in Chinese Hamster Ovary Cells and Reveals a Reduced Mitochondria Horsepower. Metabolites 2021, 11, 344. [Google Scholar] [CrossRef]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Choi, S.Y.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: A regulatory, immunosuppressive metabolite? J. Pathol. 2013, 230, 350–355. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The Warburg Effect and the Hallmarks of Cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, F.A.; Kettunen, M.I.; Day, S.E.; Hu, D.-E.; Ardenkjær-Larsen, J.H.; Zandt, R.i.t.; Jensen, P.R.; Karlsson, M.; Golman, K.; Lerche, M.H.; et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nature 2008, 453, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Raghunand, N.; Karczmar, G.S.; Bhujwalla, Z.M. MRI of the tumor microenvironment. J. Magn. Reson. Imaging 2002, 16, 430–450. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Wang, Y.H.; Scadden, D.T. Targeting the Warburg effect for leukemia therapy: Magnitude matters. Mol. Cell. Oncol. 2015, 2, e981988. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef]
- Shanmugam, M.; McBrayer, S.K.; Rosen, S.T. Targeting the Warburg effect in hematological malignancies: From PET to therapy. Curr. Opin. Oncol. 2009, 21, 531–536. [Google Scholar] [CrossRef]
- Ma, S.; Lee, H.; Jo, W.Y.; Byun, Y.H.; Shin, K.W.; Choi, S.; Oh, H.; Park, C.K.; Park, H.P. The Warburg effect in patients with brain tumors: A comprehensive analysis of clinical significance. J. Neurooncol. 2023, 165, 219–226. [Google Scholar] [CrossRef]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.E.; Yakimova, A.O. Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing. Cancers 2021, 13, 1102. [Google Scholar] [CrossRef]
- Finley, L.W.S.; Thompson, C.B. 13—The Metabolism of Cell Growth and Proliferation. In The Molecular Basis of Cancer, 4th ed.; Mendelsohn, J., Gray, J.W., Howley, P.M., Israel, M.A., Thompson, C.B., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 191–208.e2. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Hypoxia-/HIF-1α-Driven Factors of the Tumor Microenvironment Impeding Antitumor Immune Responses and Promoting Malignant Progression. Adv. Exp. Med. Biol. 2018, 1072, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.B. Perturbation-Driven Entropy as a Source of Cancer Cell Heterogeneity. Trends Cancer 2020, 6, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Hanselmann, R.G.; Welter, C. Origin of Cancer: An Information, Energy, and Matter Disease. Front. Cell. Dev. Biol. 2016, 4, 121. [Google Scholar] [CrossRef] [PubMed]
- Knapp, J.P.; Kakish, J.E.; Bridle, B.W.; Speicher, D.J. Tumor Temperature: Friend or Foe of Virus-Based Cancer Immunotherapy. Biomedicines 2022, 10, 2024. [Google Scholar] [CrossRef]
- El-Gammal, Z.; Nasr, M.A.; Elmehrath, A.O.; Salah, R.A.; Saad, S.M.; El-Badri, N. Regulation of mitochondrial temperature in health and disease. Pflügers Arch.—Eur. J. Physiol. 2022, 474, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Werb, Z.; Lu, P. The Role of Stroma in Tumor Development. Cancer J. 2015, 21, 250–253. [Google Scholar] [CrossRef]
- Baker, S.G.; Kramer, B.S. Paradoxes in carcinogenesis: New opportunities for research directions. BMC Cancer 2007, 7, 151. [Google Scholar] [CrossRef]
- Robinson, A.J.; Jain, A.; Sherman, H.G.; Hague, R.J.M.; Rahman, R.; Sanjuan-Alberte, P.; Rawson, F.J. Toward Hijacking Bioelectricity in Cancer to Develop New Bioelectronic Medicine. Adv. Ther. 2021, 4, 2000248. [Google Scholar] [CrossRef]
- Tiwari, B.S.; Belenghi, B.; Levine, A. Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 2002, 128, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Pokorný, J.; Pokorný, J.; Borodavka, F. Warburg effect—Damping of electromagnetic oscillations. Electromagn. Biol. Med. 2017, 36, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Pokorný, J.; Pokorný, J.; Kobilková, J.; Jandová, A.; Holaj, R. Cancer Development and Damped Electromagnetic Activity. Appl. Sci. 2020, 10, 1826. [Google Scholar] [CrossRef]
- Behnam, B.; Taghizadeh-Hesary, F. Mitochondrial Metabolism: A New Dimension of Personalized Oncology. Cancers 2023, 15, 4058. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, B. (Ed.) Dynamics of water: Molecular motions and hydrogen-bond-breaking kinetics. In Water in Biological and Chemical Processes: From Structure and Dynamics to Function; Cambridge University Press: Cambridge, UK, 2013; pp. 27–60. [Google Scholar] [CrossRef]
- Marques, M.P.M.; Batista de Carvalho, A.L.M.; Mamede, A.P.; Dopplapudi, A.; García Sakai, V.; Batista de Carvalho, L.A.E. Role of intracellular water in the normal-to-cancer transition in human cells-insights from quasi-elastic neutron scattering. Struct. Dyn. 2020, 7, 054701. [Google Scholar] [CrossRef] [PubMed]
- Tanner, K.; Mori, H.; Mroue, R.; Bruni-Cardoso, A.; Bissell, M.J. Coherent angular motion in the establishment of multicellular architecture of glandular tissues. Proc. Natl. Acad. Sci. USA 2012, 109, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Cho, J.; Lee, K. Tumour Regression via Integrative Regulation of Neurological, Inflammatory, and Hypoxic Tumour Microenvironment. Biomol. Ther. 2020, 28, 119–130. [Google Scholar] [CrossRef]
- Scheid, A.D.; Beadnell, T.C.; Welch, D.R. Roles of mitochondria in the hallmarks of metastasis. Br. J. Cancer 2021, 124, 124–135. [Google Scholar] [CrossRef]
- Song, I.S.; Jeong, J.Y.; Jeong, S.H.; Kim, H.K.; Ko, K.S.; Rhee, B.D.; Kim, N.; Han, J. Mitochondria as therapeutic targets for cancer stem cells. World J. Stem Cells 2015, 7, 418–427. [Google Scholar] [CrossRef]
- Taghizadeh-Hesary, F.; Akbari, H.; Bahadori, M.; Behnam, B. Targeted Anti-Mitochondrial Therapy: The Future of Oncology. Genes 2022, 13, 1728. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. eBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell. Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef] [PubMed]
- Min, H.Y.; Lee, H.Y. Molecular targeted therapy for anticancer treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef]
- Iwasaki, H.; Suda, T. Cancer stem cells and their niche. Cancer Sci. 2009, 100, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Ansooya, B.; Patricia, S. Mitochondrial determinants of chemoresistance. Mitochondrial Determ. Chemoresistance 2019, 2, 634–646. [Google Scholar] [CrossRef]
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Mukherjee, L.; Holyoake, T.L. Concise review: Cancer cells escape from oncogene addiction: Understanding the mechanisms behind treatment failure for more effective targeting. Stem Cells 2014, 32, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Saw, K.M.; Eaves, A.; Eaves, C. Instability of BCR-ABL gene in primary and cultured chronic myeloid leukemia stem cells. J. Natl. Cancer Inst. 2007, 99, 680–693. [Google Scholar] [CrossRef]
- Ladanie, A.; Schmitt, A.M.; Speich, B.; Naudet, F.; Agarwal, A.; Pereira, T.V.; Sclafani, F.; Herbrand, A.K.; Briel, M.; Martin-Liberal, J.; et al. Clinical Trial Evidence Supporting US Food and Drug Administration Approval of Novel Cancer Therapies Between 2000 and 2016. JAMA Netw. Open 2020, 3, e2024406. [Google Scholar] [CrossRef]
- Del Paggio, J.C.; Berry, J.S.; Hopman, W.M.; Eisenhauer, E.A.; Prasad, V.; Gyawali, B.; Booth, C.M. Evolution of the Randomized Clinical Trial in the Era of Precision Oncology. JAMA Oncol. 2021, 7, 728–734. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxid. Med. Cell. Longev. 2018, 2018, 7582730. [Google Scholar] [CrossRef]
- Averbeck, D.; Rodriguez-Lafrasse, C. Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts. Int. J. Mol. Sci. 2021, 22, 11047. [Google Scholar] [CrossRef]
- Cohen, J.B.; Brown, N.J.; Brown, S.A.; Dent, S.; van Dorst, D.C.H.; Herrmann, S.M.; Lang, N.N.; Oudit, G.Y.; Touyz, R.M. Cancer Therapy-Related Hypertension: A Scientific Statement From the American Heart Association. Hypertension 2023, 80, e46–e57. [Google Scholar] [CrossRef]
- Bikomeye, J.C.; Terwoord, J.D.; Santos, J.H.; Beyer, A.M. Emerging mitochondrial signaling mechanisms in cardio-oncology: Beyond oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H702. [Google Scholar] [CrossRef]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat. 2012, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Liao, Y.; Qin, J.Y.; Li, W.H.; Tang, Z.Y. Adverse effects of chemoradiotherapy on invasion and metastasis of tumor cells. Genes Dis. 2020, 7, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Bujko, K.; Mack, A.; Kucia, M.; Ratajczak, J. Cancer from the perspective of stem cells and misappropriated tissue regeneration mechanisms. Leukemia 2018, 32, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Nice, E.C.; Huang, C.; Fu, L. Mitochondrial adaptation in cancer drug resistance: Prevalence, mechanisms, and management. J. Hematol. Oncol. 2022, 15, 97. [Google Scholar] [CrossRef]
- Bao, B.; Ahmad, A.; Azmi, A.S.; Ali, S.; Sarkar, F.H. Overview of cancer stem cells (CSCs) and mechanisms of their regulation: Implications for cancer therapy. Curr. Protoc. Pharmacol. 2013, 61, 14.25.1–14.25.14. [Google Scholar] [CrossRef]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, 1596004. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed]
- Moradi Kashkooli, F.; Soltani, M. Evaluation of solid tumor response to sequential treatment cycles via a new computational hybrid approach. Sci. Rep. 2021, 11, 21475. [Google Scholar] [CrossRef] [PubMed]
- Erdi, Y.E. Limits of Tumor Detectability in Nuclear Medicine and PET. Mol. Imaging Radionucl. Ther. 2012, 21, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A. Disappearing breast cancers. Curr. Oncol. 2012, 19, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.M.; Olsen, M.W.; Ley, C.D.; Klausen, T.L.; Mortensen, J.; Højgaard, L.; Kristjansen, P.E. How few cancer cells can be detected by positron emission tomography? A frequent question addressed by an in vitro study. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.X.; Chen, J.J.; Sun, Y.B.; Chen, T.Q.; Wang, J.; Yu, S.C. Mitochondria in cancer stem cells: Achilles heel or hard armor. Trends Cell Biol. 2023, 33, 708–727. [Google Scholar] [CrossRef]
- Vuda, M.; Kamath, A. Drug induced mitochondrial dysfunction: Mechanisms and adverse clinical consequences. Mitochondrion 2016, 31, 63–74. [Google Scholar] [CrossRef]
- Fromenty, B.; Pessayre, D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Moen, I.; Stuhr, L.E. Hyperbaric oxygen therapy and cancer—A review. Target. Oncol. 2012, 7, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Martella, R.; Ravera, S.; Marini, C.; Capitanio, S.; Orengo, A.; Emionite, L.; Lavarello, C.; Amaro, A.; Petretto, A.; et al. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget 2015, 6, 11806–11819. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, P.; Baghli, I.; Gourjon, G.; Seyfried, T.N. Mitochondrial–Stem Cell Connection: Providing Additional Explanations for Understanding Cancer. Metabolites 2024, 14, 229. https://doi.org/10.3390/metabo14040229
Martinez P, Baghli I, Gourjon G, Seyfried TN. Mitochondrial–Stem Cell Connection: Providing Additional Explanations for Understanding Cancer. Metabolites. 2024; 14(4):229. https://doi.org/10.3390/metabo14040229
Chicago/Turabian StyleMartinez, Pierrick, Ilyes Baghli, Géraud Gourjon, and Thomas N. Seyfried. 2024. "Mitochondrial–Stem Cell Connection: Providing Additional Explanations for Understanding Cancer" Metabolites 14, no. 4: 229. https://doi.org/10.3390/metabo14040229
APA StyleMartinez, P., Baghli, I., Gourjon, G., & Seyfried, T. N. (2024). Mitochondrial–Stem Cell Connection: Providing Additional Explanations for Understanding Cancer. Metabolites, 14(4), 229. https://doi.org/10.3390/metabo14040229