
Unraveling Protein-Metabolite Interactions in Precision Nutrition: A Case Study of Blueberry-Derived Metabolites Using Advanced Computational Methods

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Data Collection
2.2. Molecular Clustering
2.3. Bioactivity Prediction
2.4. Protein Target Prediction
2.5. Metabolic Pathway and Interaction Network
2.6. Molecular Docking
2.7. Molecular Dynamics (MD) Simulation
3. Results and Discussion
3.1. Molecular Clustering
3.2. Bioactivity Scores Calculation
3.3. Protein Target Prediction
3.4. Metabolic Pathway and Interaction Network Analysis
3.5. Molecular Docking
3.6. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef] [PubMed]
- Clish, C.B. Metabolomics: An Emerging but Powerful Tool for Precision Medicine. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Z.; Begley, P.; Mullard, G.; Hollywood, K.A.; Bishop, P.N. Introduction to Metabolomics and Its Applications in Ophthalmology. Eye 2016, 30, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.I.; Dunn, W.B.; Griffin, J.L.; Allwood, J.W.; Goodacre, R. Metabolic Fingerprinting as a Diagnostic Tool. Pharmacogenomics 2007, 8, 1243–1266. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Souza, A.L.; Gerszten, R.E.; Clish, C.B. Targeted Metabolomics. Curr. Protoc. Mol. Biol. 2012, 98. [Google Scholar] [CrossRef] [PubMed]
- Dudley, E.; Yousef, M.; Wang, Y.; Griffiths, W.J. Targeted Metabolomics and Mass Spectrometry. Adv. Protein Chem. Struct. Biol. 2010, 80, 45–83. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.; Xie, Y.; Tang, S.; Zhang, A. Small Molecule Metabolites: Discovery of Biomarkers and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Cai, Y.; Wang, Z.; Xie, Y.; Zhang, A. Decoding Functional Significance of Small Molecule Metabolites. Biomed. Pharmacother. 2023, 158, 114188. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Keshari, K.R. Metabolic Analysis as a Driver for Discovery, Diagnosis, and Therapy. Cell 2022, 185, 2678–2689. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Chen, J.-R.; Zhao, H.; Wankhade, U.D.; Chintapalli, S.V.; Li, C.; Gai, D.; Shankar, K.; Zhan, F.; Lazarenko, O.P. GPR109A Mediates the Effects of Hippuric Acid on Regulating Osteoclastogenesis and Bone Resorption in Mice. Commun. Biol. 2021, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Lazarenko, O.P.; Zhang, J.; Blackburn, M.L.; Ronis, M.J.J.; Badger, T.M. Diet-Derived Phenolic Acids Regulate Osteoblast and Adipocyte Lineage Commitment and Differentiation in Young Mice. J. Bone Miner. Res. 2014, 29, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-R.; Lazarenko, O.P.; Wu, X.; Kang, J.; Blackburn, M.L.; Shankar, K.; Badger, T.M.; Ronis, M.J.J. Dietary-Induced Serum Phenolic Acids Promote Bone Growth via P38 MAPK/β-Catenin Canonical Wnt Signaling. J. Bone Miner. Res. 2010, 25, 2399–2411. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.; Kachhap, S.; Madhukar, G.; Adepu, K.K.; Anishkin, A.; Chen, J.R.; Chintapalli, S.V. Exploring GPR109A Receptor Interaction with Hippuric Acid Using MD Simulations and CD Spectroscopy. Int. J. Mol. Sci. 2022, 23, 14778. [Google Scholar] [CrossRef] [PubMed]
- Curtis, P.J.; Berends, L.; van der Velpen, V.; Jennings, A.; Haag, L.; Chandra, P.; Kay, C.D.; Rimm, E.B.; Cassidy, A. Blueberry Anthocyanin Intake Attenuates the Postprandial Cardiometabolic Effect of an Energy-Dense Food Challenge: Results from a Double Blind, Randomized Controlled Trial in Metabolic Syndrome Participants. Clin. Nutr. 2022, 41, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Curtis, P.J.; van der Velpen, V.; Berends, L.; Jennings, A.; Feelisch, M.; Umpleby, A.M.; Evans, M.; Fernandez, B.O.; Meiss, M.S.; Minnion, M.; et al. Blueberries Improve Biomarkers of Cardiometabolic Function in Participants with Metabolic Syndrome—Results from a 6-Month, Double-Blind, Randomized Controlled Trial. Am. J. Clin. Nutr. 2019, 109, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Chen, J.; Swamidass, S.J.; Dou, Y.; Bruand, J.; Baldi, P. ChemDB: A Public Database of Small Molecules and Related Chemoinformatics Resources. Bioinformatics 2005, 21, 4133–4139. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Leite, T.B.; Gomes, D.; Miteva, M.A.; Chomilier, J.; Villoutreix, B.O.; Tuffery, P. Frog: A FRee Online DruG 3D Conformation Generator. Nucleic Acids Res. 2007, 35, W568–W572. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. In Protein Engineering. Methods in Molecular Biology; Bornscheuer, U., Höhne, M., Eds.; Humana Press: New York, NY, USA, 2018; pp. 43–67. [Google Scholar]
- Prada Gori, D.N.; Llanos, M.A.; Bellera, C.L.; Talevi, A.; Alberca, L.N. IRaPCA and SOMoC: Development and Validation of Web Applications for New Approaches for the Clustering of Small Molecules. J. Chem. Inf. Model. 2022, 62, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, Z.; Huo, R.; Cui, Z. Preparation of an Indicator Film Based on Pectin, Sodium Alginate, and Xanthan Gum Containing Blueberry Anthocyanin Extract and Its Application in Blueberry Freshness Monitoring. Heliyon 2023, 9, e14421. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C.; Gillitt, N.D.; Chen, G.-Y.; Zhang, Q.; Sha, W.; Kay, C.D.; Chandra, P.; Kay, K.L.; Lila, M.A. Blueberry and/or Banana Consumption Mitigate Arachidonic, Cytochrome P450 Oxylipin Generation During Recovery from 75-Km Cycling: A Randomized Trial. Front. Nutr. 2020, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A Web-Accessible Database of Experimentally Determined Protein-Ligand Binding Affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction Networks of Chemicals and Proteins. Nucleic Acids Res. 2007, 36, D684–D688. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Adepu, K.K.; Kachhap, S.; Anishkin, A.; Chintapalli, S.V. Structural and Energetic Insights into the Interaction of Niacin with the GPR109A Receptor. Bioinform. Biol. Insights 2021, 15, 11779322211056122. [Google Scholar] [CrossRef] [PubMed]
- Adepu, K.K.; Bhandari, D.; Anishkin, A.; Adams, S.H.; Chintapalli, S.V. Myoglobin–Pyruvate Interactions: Binding Thermodynamics, Structure–Function Relationships, and Impact on Oxygen Release Kinetics. Int. J. Mol. Sci. 2022, 23, 8766. [Google Scholar] [CrossRef] [PubMed]
- Adepu, K.K.; Bhandari, D.; Anishkin, A.; Adams, S.H.; Chintapalli, S.V. Myoglobin Interaction with Lactate Rapidly Releases Oxygen: Studies on Binding Thermodynamics, Spectroscopy, and Oxygen Kinetics. Int. J. Mol. Sci. 2022, 23, 4747. [Google Scholar] [CrossRef] [PubMed]
- Watts, F.M.; Pouland, T.; Bunce, R.A.; Berlin, K.D.; Benbrook, D.M.; Mashayekhi, M.; Bhandari, D.; Zhou, D. Activity of Oxygen-versus Sulfur-Containing Analogs of the Flex-Het Anticancer Agent SHetA2. Eur. J. Med. Chem. 2018, 158, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Chintapalli, S.V.; Jayanthi, S.; Mallipeddi, P.L.; Gundampati, R.; Suresh Kumar, T.K.; Van Rossum, D.B.; Anishkin, A.; Adams, S.H. Novel Molecular Interactions of Acylcarnitines and Fatty Acids with Myoglobin. J. Biol. Chem. 2016, 291, 25133–25143. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Hernández-Hernández, S.; Ballester, P.J. On the Best Way to Cluster NCI-60 Molecules. Biomolecules 2023, 13, 498. [Google Scholar] [CrossRef]
- Illing, E.A.; Cho, D.-Y.; Zhang, S.; Skinner, D.F.; Dunlap, Q.A.; Sorscher, E.J.; Woodworth, B.A. Chlorogenic Acid Activates CFTR-Mediated Cl- Secretion in Mice and Humans: Therapeutic Implications for Chronic Rhinosinusitis. Otolaryngol. Head. Neck Surg. 2015, 153, 291–297. [Google Scholar] [CrossRef] [PubMed]
- GULCIN, I. Antioxidant Activity of Caffeic Acid (3,4-Dihydroxycinnamic Acid). Toxicology 2006, 217, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Agunloye, O.M.; Adefegha, S.A.; Akinyemi, A.J.; Ademiluyi, A.O. Caffeic and Chlorogenic Acids Inhibit Key Enzymes Linked to Type 2 Diabetes (in Vitro): A Comparative Study. J. Basic Clin. Physiol. Pharmacol. 2015, 26, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Giresha, A.S.; Urs, D.; Pundalik, S.; Meti, R.S.; Pramod, S.N.; Supreetha, B.H.; Somegowda, M.; Dharmappa, K.K.; El-Shehawi, A.M.; Albogami, S.; et al. Sinapicacid Inhibits Group IIA Secretory Phospholipase A2 and Its Inflammatory Response in Mice. Antioxidants 2022, 11, 1251. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic Anhydrases: Novel Therapeutic Applications for Inhibitors and Activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Temperini, C.; Innocenti, A.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Carbonic Anhydrase Activators: L-Adrenaline Plugs the Active Site Entrance of Isozyme II, Activating Better Isoforms I, IV, VA, VII, and XIV. Bioorg. Med. Chem. Lett. 2007, 17, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.L.; McKenna, R. Carbonic Anhydrase II in Complex with Carboxylic Acid-Based Inhibitors. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2019, 75, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Monti, S.M.; Truppo, E.; Pedone, C.; Supuran, C.T.; De Simone, G. The First Example of a Significant Active Site Conformational Rearrangement in a Carbonic Anhydrase-Inhibitor Adduct: The Carbonic Anhydrase I–Topiramate Complex. Org. Biomol. Chem. 2010, 8, 3528. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Alterio, V.; Bua, S.; Micheli, L.; Esposito, D.; Buonanno, M.; Bartolucci, G.; Osman, S.M.; ALOthman, Z.A.; Cirilli, R.; et al. Phenyl(Thio)Phosphon(amid)Ate Benzenesulfonamides as Potent and Selective Inhibitors of Human Carbonic Anhydrases II and VII Counteract Allodynia in a Mouse Model of Oxaliplatin-Induced Neuropathy. J. Med. Chem. 2020, 63, 5185–5200. [Google Scholar] [CrossRef]
- Waheed, A.; Sly, W.S. Carbonic Anhydrase XII Functions in Health and Disease. Gene 2017, 623, 33–40. [Google Scholar] [CrossRef]
- Alterio, V.; Pan, P.; Parkkila, S.; Buonanno, M.; Supuran, C.T.; Monti, S.M.; De Simone, G. The Structural Comparison between Membrane-associated Human Carbonic Anhydrases Provides Insights into Drug Design of Selective Inhibitors. Biopolymers 2014, 101, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.M.; Tu, C.; Fisher, S.Z.; An, H.; Yoshioka, C.; Govindasamy, L.; Laipis, P.J.; Agbandje-McKenna, M.; Silverman, D.N.; McKenna, R. Human Carbonic Anhydrase III: Structural and Kinetic Study of Catalysis and Proton Transfer. Biochemistry 2005, 44, 10046–10053. [Google Scholar] [CrossRef] [PubMed]
- Mickevičiūtė, A.; Timm, D.D.; Gedgaudas, M.; Linkuvienė, V.; Chen, Z.; Waheed, A.; Michailovienė, V.; Zubrienė, A.; Smirnov, A.; Čapkauskaitė, E.; et al. Intrinsic Thermodynamics of High Affinity Inhibitor Binding to Recombinant Human Carbonic Anhydrase IV. Eur. Biophys. J. 2018, 47, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Kazokaitė, J.; Kairys, V.; Smirnovienė, J.; Smirnov, A.; Manakova, E.; Tolvanen, M.; Parkkila, S.; Matulis, D. Engineered Carbonic Anhydrase VI-Mimic Enzyme Switched the Structure and Affinities of Inhibitors. Sci. Rep. 2019, 9, 12710. [Google Scholar] [CrossRef] [PubMed]
- Zakšauskas, A.; Čapkauskaitė, E.; Paketurytė-Latvė, V.; Smirnov, A.; Leitans, J.; Kazaks, A.; Dvinskis, E.; Stančaitis, L.; Mickevičiūtė, A.; Jachno, J.; et al. Methyl 2-Halo-4-Substituted-5-Sulfamoyl-Benzoates as High Affinity and Selective Inhibitors of Carbonic Anhydrase IX. Int. J. Mol. Sci. 2021, 23, 130. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, A.; Monti, S.M.; Hilvo, M.; Parkkila, S.; Romano, V.; Scaloni, A.; Pedone, C.; Scozzafava, A.; Supuran, C.T.; De Simone, G. Crystal Structure of Human Carbonic Anhydrase XIII and Its Complex with the Inhibitor Acetazolamide. Proteins Struct. Funct. Bioinforma. 2009, 74, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-K.; Boron, W.F.; Occhipinti, R. Potential Novel Role of Membrane-Associated Carbonic Anhydrases in the Kidney. Int. J. Mol. Sci. 2023, 24, 4251. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, K.C.; Guex, C.G.; Graiczik, J.; Reginato, F.Z.; Engelmann, A.M.; De Andrade, C.M.; Timmers, L.F.S.M.; Bauermann, L.D.F. Caffeic Acid and Ferulic Acid Can Improve Toxicological Damage Caused by Iron Overload Mediated by Carbonic Anhydrase Inhibition. Drug Chem. Toxicol. 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gülçin, İ.; Scozzafava, A.; Supuran, C.T.; Akıncıoğlu, H.; Koksal, Z.; Turkan, F.; Alwasel, S. The Effect of Caffeic Acid Phenethyl Ester (CAPE) on Metabolic Enzymes Including Acetylcholinesterase, Butyrylcholinesterase, Glutathione S-Transferase, Lactoperoxidase, and Carbonic Anhydrase Isoenzymes I, II, IX, and XII. J. Enzyme Inhib. Med. Chem. 2016, 31, 1095–1101. [Google Scholar] [CrossRef]
- Karpuzoglu, E.; Holladay, S.D.; Gogal, R.M. Parabens: Potential Impact of Low-Affinity Estrogen Receptor Binding Chemicals on Human Health. J. Toxicol. Environ. Heal. Part B 2013, 16, 321–335. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Marciniak, B.; Drozda, R.; Kontek, R. Targeting Carbonic Anhydrase IX and XII Isoforms with Small Molecule Inhibitors and Monoclonal Antibodies. J. Enzyme Inhib. Med. Chem. 2022, 37, 1278–1298. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Ye, H.; Zhang, J.; Ren, L. Recent Advances in Nuclear Receptors-Mediated Health Benefits of Blueberry. Phytomedicine 2022, 100, 154063. [Google Scholar] [CrossRef] [PubMed]
- Taheri, Y.; Suleria, H.A.R.; Martins, N.; Sytar, O.; Beyatli, A.; Yeskaliyeva, B.; Seitimova, G.; Salehi, B.; Semwal, P.; Painuli, S.; et al. Myricetin Bioactive Effects: Moving from Preclinical Evidence to Potential Clinical Applications. BMC Complement. Med. Ther. 2020, 20, 241. [Google Scholar] [CrossRef]
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the Development of Human Carbonic Anhydrase Inhibitors and Their Pharmacological Applications: Where Are We Today? Med. Res. Rev. 2020, 40, 2485–2565. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Vyas, P.; Weber, J.T. Biochemical Properties and Neuroprotective Effects of Compounds in Various Species of Berries. Molecules 2017, 23, 26. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Cerri, C.; Nencetti, S.; Orlandini, E. Carbonic Anhydrase Inhibitors and Epilepsy: State of the Art and Future Perspectives. Molecules 2021, 26, 6380. [Google Scholar] [CrossRef] [PubMed]
- García-Llorca, A.; Carta, F.; Supuran, C.T.; Eysteinsson, T. Carbonic Anhydrase, Its Inhibitors and Vascular Function. Front. Mol. Biosci. 2024, 11, 1338528. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, A.P.; Ciampa, A.; Ingallina, C.; Mannina, L.; Capitani, D.; Ernesti, I.; Maggi, E.; Businaro, R.; Del Ben, M.; Engel, P.; et al. Blueberry-Based Meals for Obese Patients with Metabolic Syndrome: A Multidisciplinary Metabolomic Pilot Study. Metabolites 2019, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.H.; Selemidis, S.; Bozinovski, S.; Vlahos, R. Pathobiological Mechanisms Underlying Metabolic Syndrome (MetS) in Chronic Obstructive Pulmonary Disease (COPD): Clinical Significance and Therapeutic Strategies. Pharmacol. Ther. 2019, 198, 160–188. [Google Scholar] [CrossRef]
- de Oliveira, M.S.; Pellenz, F.M.; de Souza, B.M.; Crispim, D. Blueberry Consumption and Changes in Obesity and Diabetes Mellitus Outcomes: A Systematic Review. Metabolites 2022, 13, 19. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Djoumbou-Feunang, Y.; Fiamoncini, J.; Gil-de-la-Fuente, A.; Greiner, R.; Manach, C.; Wishart, D.S. BioTransformer: A Comprehensive Computational Tool for Small Molecule Metabolism Prediction and Metabolite Identification. J. Cheminform. 2019, 11, 2. [Google Scholar] [CrossRef] [PubMed]




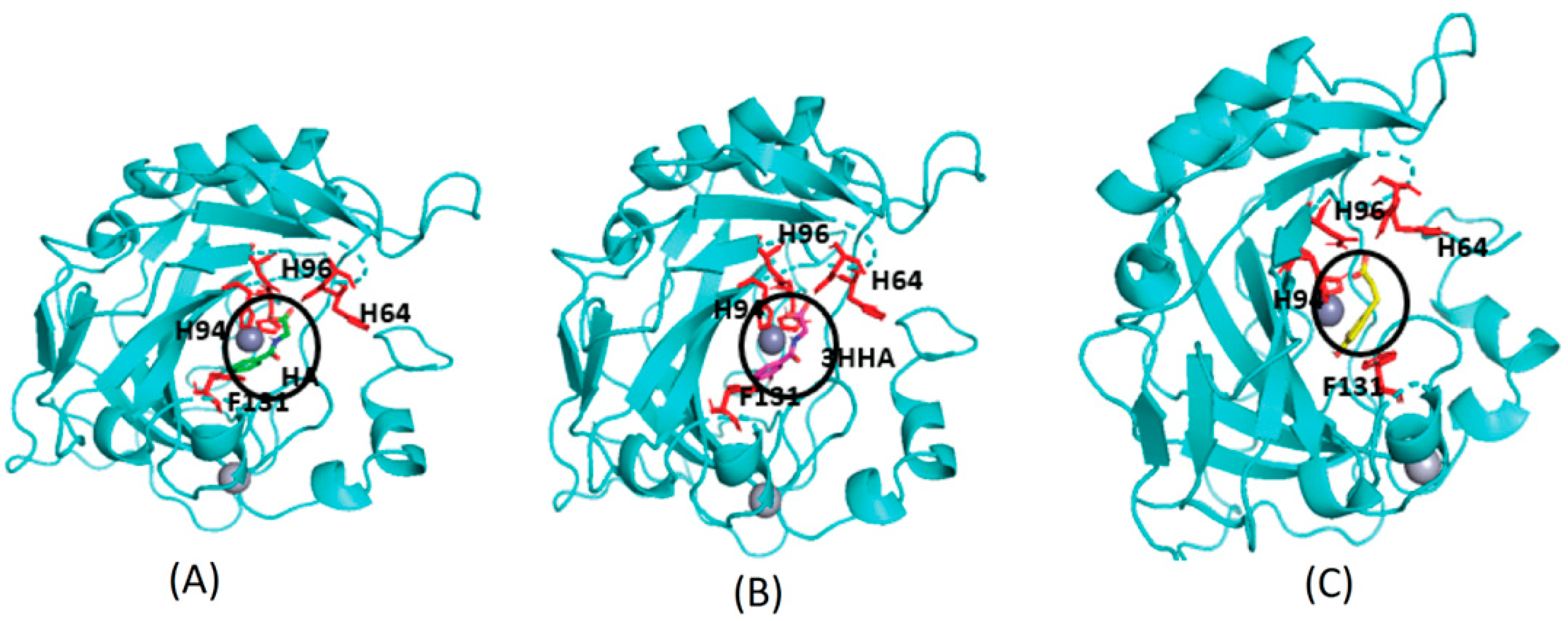

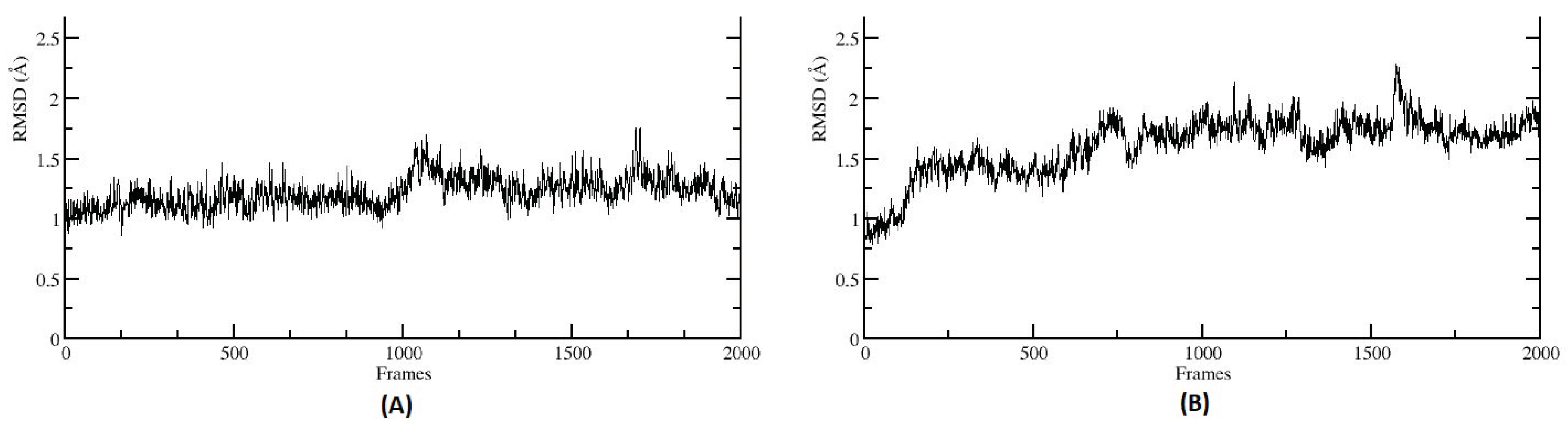


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds (Cluster-0) | GPCR Ligand Activity | Channel Activity | Kinase Inhibitor Activity | Protease Inhibitor Activity | Enzyme Inhibitor Activity | Nuclear Receptor Ligand Activity | Protein Target (90–100% Confidence Hit) |
|---|---|---|---|---|---|---|---|
| 4-Hydroxybenzoic acid | −0.98 | −0.93 | −1.21 | −1.19 | −0.41 | −0.62 | CA I, CA II, CA III, CA IV, (D1A), EGFR |
| 3,4-Dihydroxyhydrocinnamic acid | −0.29 | −0.06 | −0.79 | −0.52 | 0.01 | −0.12 | - |
| 3-(4-Hydroxyphenyl) propionic acid (Desaminotyrosine) | −0.35 | −0.07 | −0.89 | −0.57 | −0.04 | −0.14 | - |
| 3,4-Dihydroxybenzeneacetic acid | −0.46 | −0.06 | −0.92 | −0.64 | −0.04 | −0.16 | - |
| Hippuric acid | −0.52 | −0.2 | −0.87 | −0.39 | −0.19 | −0.76 | CA II, CA III, CA IX, HCAR2 |
| 3,5-Dihydroxybenzoic acid | −0.86 | −0.34 | −1.04 | −1.08 | −0.31 | −0.47 | CA I, CA II, CA VII, CA XII, CA XIV, CA IX |
| 3,4-dihydroxybenzoic acid (Protocatechuic acid) | −0.88 | −0.35 | −1.1 | −1.09 | −0.34 | −0.58 | CA I, CA II, CA VII, CA VI, CA XII, CA XIV, CA IX, CA IV |
| 2,4-Dihydroxybenzoic acid | −0.81 | −0.33 | −0.99 | −1.02 | −0.28 | −0.5 | CA I, CA II, CA XII |
| 4-Hydroxycinnamic acid | −0.56 | −0.26 | −0.91 | −0.87 | −0.15 | −0.12 | AR, CA I, CA II, CA III, CA VI, CA VII, CA XII, ERβ |
| 2,3-Dihydroxybenzoic acid (2-Pyrocatechuic acid) | −0.8 | −0.24 | −1.02 | −1.06 | −0.3 | −0.74 | - |
| 2,4,6-Trihydroxybenzaldehyde | −0.96 | −0.3 | −0.82 | −1.33 | −0.46 | −0.59 | - |
| 2-Hydroxybenzoic acid (Salicylic acid) | −0.98 | −0.43 | −1.22 | −1.14 | −0.41 | −0.79 | CA I, CA II, CA XII |
| 3-Hydroxybenzoic acid | −0.99 | −0.42 | −1.25 | −1.21 | −0.43 | −0.61 | CA I, CA II, CA VI, CA XII, CA IX, HCAR1, HCAR2 |
| 3-Hydroxyhippuric acid | −0.37 | −0.12 | −0.7 | −0.33 | −0.07 | −0.42 | - |
| 4-Hydroxybenzyl alcohol | −1.91 | −1.2 | −2.02 | −2.02 | −1.31 | −1.78 | - |
| Compounds (Cluster-1) | |||||||
| 4-Hydroxy-3-methoxyphenylacetic acid (Homovanillic acid) | −0.65 | −0.28 | −0.69 | −0.82 | −0.15 | −0.44 | - |
| 4-Hydroxy-3,5-dimethoxybenzoic acid (Syringic acid) | −0.29 | −0.14 | 0 | −0.27 | −0.62 | −0.74 | CA I, CA II, CA III, CA VII |
| Chlorogenic acid (3-Caffeoylquinic acid) | −0.41 | −0.24 | −0.93 | −0.59 | −0.16 | −0.29 | - |
| 3-Methoxybenzenepropanoic acid | −0.29 | −0.14 | −0.7 | −0.56 | −0.03 | −0.16 | - |
| 3-(3-hydroxy-4-methoxyphenyl) propanoic acid | −0.47 | −0.3 | −0.72 | −0.81 | −0.12 | −0.14 | - |
| 4-Hydroxy-3-methoxycinnamic acid (Ferulic acid) | −0.71 | −0.32 | −0.82 | −0.95 | −0.25 | −0.56 | CA II, CA VII |
| 3,5-Dihydroxy-4-methoxybenzoic acid (4-O-Methylgallic acid) | −0.29 | −0.14 | −0.7 | −0.56 | −0.03 | −0.16 | - |
| 3-(4-Hydroxy-3-methoxyphenyl) propanoic acid (Hydroferulic acid) | −0.85 | −0.42 | −0.99 | −1.12 | −0.35 | −0.61 | - |
| 3-Hydroxy-4-methoxybenzoic acid | −0.32 | −0.2 | −0.47 | −0.56 | 0.03 | −0.03 | - |
| 4-Hydroxy-3,5-dimethoxycinnamic acid (Sinapic acid) | −0.85 | −0.42 | −0.99 | −1.12 | −0.35 | −0.61 | - |
| 4-Hydroxy-3-methoxybenzoic acid (Vanillic acid) | −1.13 | −0.51 | −1.22 | −1.31 | −0.62 | −0.88 | - |
| 4-Hydroxybenzoic acid methyl ester (Methylparaben) | −0.23 | −0.01 | −0.78 | −0.53 | 0.1 | −0.08 | CA VII, CAXII |
| Compounds | CA-I | CA-II | CA-III | CA-IV | CA-VA | CA-VI | CA-VII | CA-IX | CA-XII | CA-XIII | CA-XIV |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Adrenaline | −6.2 | −5.8 | −5.7 | −5.5 | −6.1 | −6.1 | −6.1 | −6.4 | −6 | −5.7 | −5.9 |
| Dopa | −6.6 | −6.3 | −6.5 | −5.5 | −6.6 | −6.5 | −6.5 | −6.9 | −6.3 | −5.9 | −6.2 |
| Dopamine | −6.2 | −5.6 | −5.5 | −5.2 | −6.1 | −6 | −6.2 | −6.2 | −5.8 | −5.5 | −6 |
| Histamine | −4.5 | −4.5 | −3.9 | −4.2 | −4.1 | −4.7 | −4.3 | −4.8 | −4.4 | −4.3 | −4.3 |
| Histidine | −5.3 | −5.1 | −5 | −5 | −5.3 | −5.3 | −5.4 | −5.8 | −5.2 | −5.3 | −5.3 |
| Phenylalanine | −5.8 | −6.1 | −6.3 | −5.6 | −5.7 | −6 | −5.8 | −6.3 | −6.2 | −5.5 | −6.4 |
| Serotonine | −6.1 | −6.1 | −5.5 | −5.4 | −6 | −6.4 | −6.1 | −7 | −6.2 | −5.8 | −6.1 |
| Tryptophan | −6.4 | −6.6 | −6.4 | −6.1 | −6.6 | −7 | −6.6 | −7.2 | −6.8 | −6.3 | −6.7 |
| Tyrosine | −6.1 | −6 | −6.2 | −5.5 | −6 | −6 | −5.8 | −6.7 | −6 | −5.7 | −6.2 |
| Acetazolamide | −6.1 | −6.2 | −5.6 | −5.6 | −6.3 | −6.7 | −6.1 | −6.3 | −5.8 | −5.9 | −6.2 |
| Brinzolamide | −6.2 | −7.1 | −6.0 | −6.2 | −6.7 | −6.9 | −6.6 | −6.4 | −6.3 | −6.5 | −6.9 |
| Celecoxib | −7.8 | −8.2 | −7.8 | −7.8 | −8.8 | −8.6 | −7.5 | −8.3 | −8.9 | −8.0 | −8.4 |
| COUMATE | −7.0 | −7.6 | −7.0 | −6.7 | −7.2 | −7.5 | −7.4 | −7.4 | −7.2 | −6.5 | −7.5 |
| Dichlorophenamide | −6.3 | −6.6 | −6.2 | −6.6 | −7.1 | −6.7 | −6.2 | −7.0 | −6.8 | −5.8 | −7.0 |
| Dorzolamide | −6.5 | −6.3 | −6.8 | −6.5 | −7.6 | −7.6 | −6.7 | −6.8 | −7.0 | −6.6 | −7.7 |
| EMATE | −8.5 | −8.3 | −7.5 | −7.6 | −8.1 | −8.4 | −8.1 | −8.5 | −7.4 | −7.5 | −8.1 |
| Ethoxzolamide | −6.1 | −6.1 | −5.9 | −5.8 | −6.4 | −6.5 | −6.6 | −6.8 | −6.1 | −5.9 | −6.4 |
| Indisulam | −7.9 | −8.5 | −7.4 | −7.6 | −8.3 | −9.2 | −8.1 | −8.9 | −7.8 | 7.5 | −8.7 |
| Methazolamide | −5.6 | −6.4 | −5.6 | −5.7 | −6.4 | −6.4 | −6.2 | −6.4 | −6.6 | −5.6 | −6.3 |
| Saccharin | −6.4 | −6.3 | −5.9 | −6.3 | −6.9 | −6.8 | −6.8 | −7.0 | −6.2 | −5.8 | −7.1 |
| Sulpiride | −7.0 | −7.1 | −6.7 | −6.7 | −7.4 | −7.4 | −6.5 | −7.5 | −7.3 | −6.5 | −7.4 |
| Sulthiame | −7.5 | −7.1 | −6.6 | −6.4 | −6.8 | −7.6 | −7.2 | −7.3 | −7.2 | −6.1 | −7.3 |
| Topiramate | −7.1 | −7.2 | −7.2 | −6.7 | −6.8 | −7.0 | −6.3 | −7.9 | −7.6 | −6.6 | −7.5 |
| Valdecoxib | −7.3 | −8.2 | −7.7 | −7.7 | −7.7 | −8.7 | −7.4 | −8.2 | −8.3 | −8.0 | −8.2 |
| Zonisamide | −6.9 | −6.9 | −6.8 | −6.4 | −6.6 | −7.1 | −7.0 | −6.9 | −6.6 | −6.0 | −7.0 |
| Cluster-0 Metabolites | CA I | CA II | CA III | CA IV | CA VA | CA VI | CA VII | CA IX | CA XII | CA XIII | CA XIV |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2,4,6-trihydroxybenzaldehyde | −5.6 | −5.7 | −5.2 | −5.2 | −5.4 | −5.5 | −5.6 | −6.7 | −5.2 | −4.9 | −5.9 |
| 2,4-Dihydroxybenzoic acid | −6.1 | −6.2 | −5.4 | −5.4 | −5.8 | −6.2 | −6.3 | −6.8 | −5.7 | −5.4 | −5.9 |
| 2,3-Dihydroxybenzoic acid (2-Pyrocatechuic acid) | −6.3 | −6.1 | −6.0 | −5.5 | −6.0 | −6.4 | −6.5 | −6.8 | −5.8 | −5.6 | −6.0 |
| 3,4-Dihydroxybenzeneacetic acid | −6.4 | −5.9 | −5.8 | −5.4 | −6.3 | −6.1 | −6.5 | −6.6 | −6.4 | −5.7 | −6.2 |
| 3,4-Dihydroxyhydrocinnamic acid | −6.6 | −6.1 | −6.2 | −5.6 | −6.3 | −6.3 | −6.6 | −7.0 | −6.3 | −5.9 | −6.4 |
| 3,5-Dihydroxybenzoic acid | −6.1 | −5.8 | −5.4 | −5.5 | −5.8 | −6.1 | −5.7 | −6.7 | −5.6 | −5.2 | −5.6 |
| 3-Hydroxybenzoic acid | −6.0 | −5.7 | −5.8 | −5.6 | −5.8 | −6.1 | −5.9 | −6.4 | −5.7 | −5.5 | −5.8 |
| 3-Hydroxyhippuric acid | −6.3 | −6.9 | −6.8 | −6.3 | −6.8 | −6.7 | −6.3 | −7.5 | −6.4 | −6.4 | −6.5 |
| 4-Hydroxybenzoic acid | −5.8 | −5.8 | −5.3 | −5.2 | −5.6 | −5.8 | −5.4 | −6.2 | −5.2 | −5.3 | −5.6 |
| 4-Hydroxybenzyl alcohol | −5.4 | −5.3 | −5.2 | −4.8 | −5.3 | −5.2 | −5.1 | −5.9 | −4.9 | −5.0 | −5.0 |
| 4-Hydroxycinnamic acid | −5.9 | −5.7 | −6.1 | −5.8 | −5.7 | −5.8 | −5.7 | −7.2 | −5.9 | −5.9 | −6.0 |
| 3-(4-Hydroxyphenyl) propionic acid (Desaminotyrosine) | −6.1 | −5.7 | −5.9 | −5.4 | −5.7 | −6.1 | −5.7 | −6.9 | −5.8 | −5.7 | −6.0 |
| 2-Benzamidoacetic acid (Hippuric acid) | −5.9 | −6.6 | −6.7 | −6.1 | −6.6 | −6.6 | −6.0 | −6.7 | −6.2 | −6.2 | −6.5 |
| 3,4-Dihydroxybenzoic acid (Protocatechuic acid) | −6.4 | −6.1 | −5.6 | −5.4 | −6.3 | −6.5 | −6.4 | −6.4 | −6.1 | −5.7 | −6.1 |
| 2-Hydroxybenzoic acid (Salicylic acid) | −5.9 | −6.0 | −5.8 | −5.5 | −5.9 | −6.2 | −6.3 | −6.3 | −5.6 | −5.4 | −5.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhandari, D.; Adepu, K.K.; Anishkin, A.; Kay, C.D.; Young, E.E.; Baumbauer, K.M.; Ghosh, A.; Chintapalli, S.V. Unraveling Protein-Metabolite Interactions in Precision Nutrition: A Case Study of Blueberry-Derived Metabolites Using Advanced Computational Methods. Metabolites 2024, 14, 430. https://doi.org/10.3390/metabo14080430
Bhandari D, Adepu KK, Anishkin A, Kay CD, Young EE, Baumbauer KM, Ghosh A, Chintapalli SV. Unraveling Protein-Metabolite Interactions in Precision Nutrition: A Case Study of Blueberry-Derived Metabolites Using Advanced Computational Methods. Metabolites. 2024; 14(8):430. https://doi.org/10.3390/metabo14080430
Chicago/Turabian StyleBhandari, Dipendra, Kiran Kumar Adepu, Andriy Anishkin, Colin D. Kay, Erin E. Young, Kyle M. Baumbauer, Anuradha Ghosh, and Sree V. Chintapalli. 2024. "Unraveling Protein-Metabolite Interactions in Precision Nutrition: A Case Study of Blueberry-Derived Metabolites Using Advanced Computational Methods" Metabolites 14, no. 8: 430. https://doi.org/10.3390/metabo14080430