

Challenges in Lipidomics Biomarker Identification: Avoiding the Pitfalls and Improving Reproducibility
 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. PANC-1 Lipid Extraction LC–MS Dataset
2.2. Comparison of Outputs
2.3. Post-Software Quality Control Checks of Data
3. Results
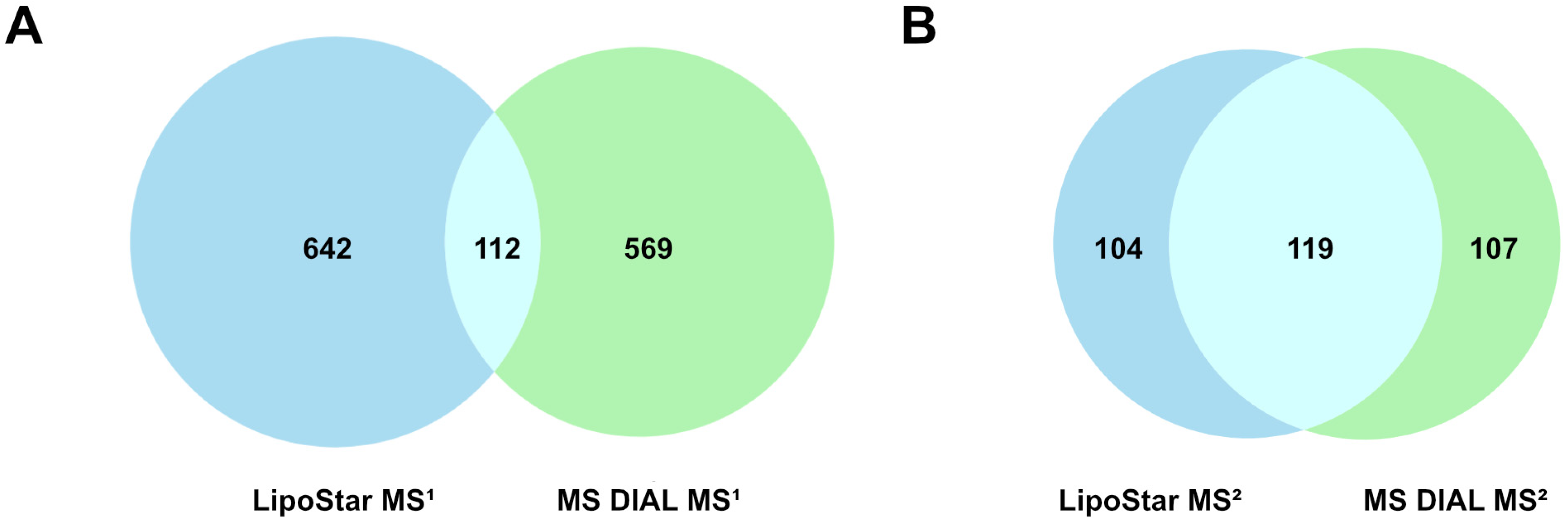
3.1. Comparison of MS DIAL and Lipostar Outputs
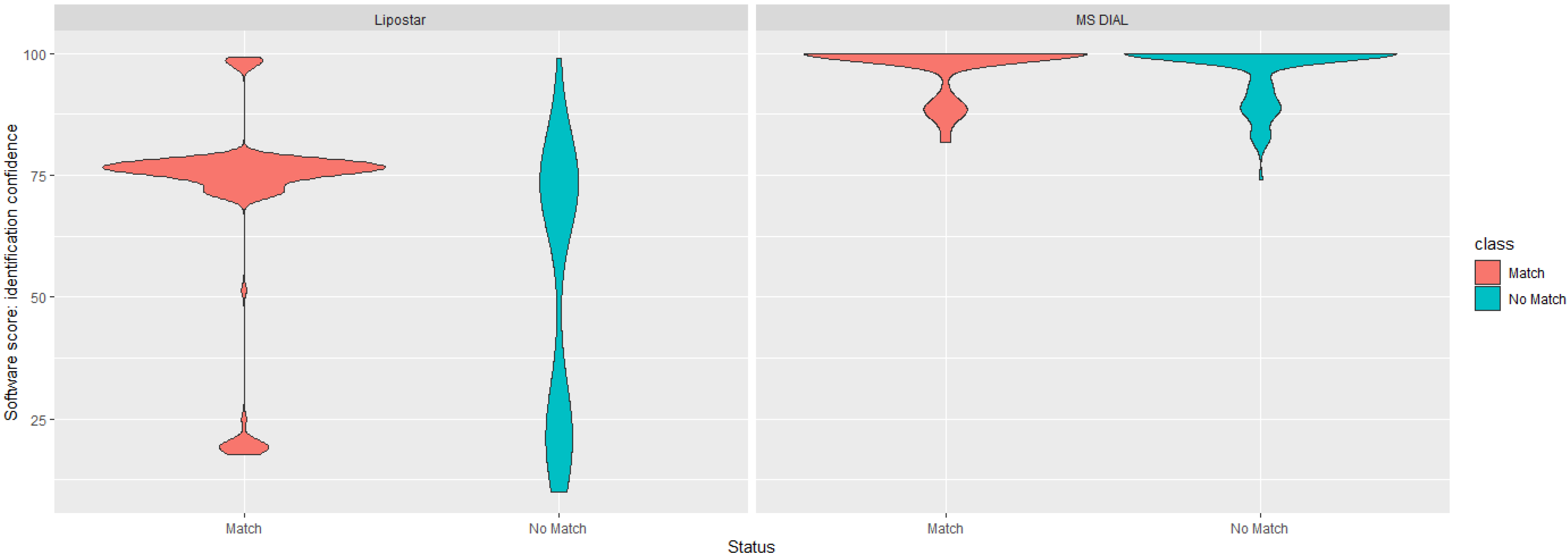
3.2. Data-Driven Investigation of Putative Lipid Identifications
3.3. Manual Investigation of Putative Lipid Identifications
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wenk, M.R. The Emerging Field of Lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Han, X. Lipidomics for Studying Metabolism. Nat. Rev. Endocrinol. 2016, 12, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-Omics Approaches to Disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-Omics Data Integration, Interpretation, and Its Application. Bioinform Biol. Insights 2020, 14, 117793221989905. [Google Scholar] [CrossRef] [PubMed]
- Sarmad, S.; Viant, M.R.; Dunn, W.B.; Goodacre, R.; Wilson, I.D.; Chappell, K.E.; Griffin, J.L.; O’Donnell, V.B.; Naicker, B.; Lewis, M.R.; et al. A Proposed Framework to Evaluate the Quality and Reliability of Targeted Metabolomics Assays from the UK Consortium on Metabolic Phenotyping (MAP/UK). Nat. Protoc. 2023, 18, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Köfeler, H.C.; Ahrends, R.; Baker, E.S.; Ekroos, K.; Han, X.; Hoffmann, N.; Holčapek, M.; Wenk, M.R.; Liebisch, G. Recommendations for Good Practice in MS-Based Lipidomics. J. Lipid Res. 2021, 62, 100138. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, G.; Gika, H.; Raftery, D.; Goodacre, R.; Plumb, R.S.; Wilson, I.D. Ensuring Fact-Based Metabolite Identification in Liquid Chromatography–Mass Spectrometry-Based Metabolomics. Anal. Chem. 2023, 95, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Lipidomics Standards Initiative Consortium. Lipidomics Needs More Standardization. Nat. Metab. 2019, 1, 745–747. [Google Scholar] [CrossRef]
- Lipidomics Standards Initiative. Available online: https://lipidomicstandards.org/ (accessed on 20 June 2023).
- McDonald, J.G.; Ejsing, C.S.; Kopczynski, D.; Holčapek, M.; Aoki, J.; Arita, M.; Arita, M.; Baker, E.S.; Bertrand-Michel, J.; Bowden, J.A.; et al. Introducing the Lipidomics Minimal Reporting Checklist. Nat. Metab. 2022, 4, 1086–1088. [Google Scholar] [CrossRef]
- MSI Board Members; Sansone, S.-A.; Fan, T.; Goodacre, R.; Griffin, J.L.; Hardy, N.W.; Kaddurah-Daouk, R.; Kristal, B.S.; Lindon, J.; Mendes, P.; et al. The Metabolomics Standards Initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar] [CrossRef]
- Spicer, R.A.; Salek, R.; Steinbeck, C. A Decade after the Metabolomics Standards Initiative It’s Time for a Revision. Sci. Data 2017, 4, 170138. [Google Scholar] [CrossRef]
- Saunders, K.D.G.; Von Gerichten, J.; Lewis, H.-M.; Gupta, P.; Spick, M.; Costa, C.; Velliou, E.; Bailey, M.J. Single-Cell Lipidomics Using Analytical Flow LC-MS Characterizes the Response to Chemotherapy in Cultured Pancreatic Cancer Cells. Anal. Chem. 2023, 95, 14727–14735. [Google Scholar] [CrossRef] [PubMed]
- Avela, H.F.; Sirén, H. Advances in Lipidomics. Clin. Chim. Acta 2020, 510, 123–141. [Google Scholar] [CrossRef]
- Song, H.; Ladenson, J.; Turk, J. Algorithms for Automatic Processing of Data from Mass Spectrometric Analyses of Lipids. J. Chromatogr. B 2009, 877, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Kanu, A.B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H.H. Ion Mobility-Mass Spectrometry. J. Mass Spectrom. 2008, 43, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, E.S.; Matyushin, D.D.; Plyushchenko, I.V.; Stavrianidi, A.N.; Buryak, A.K. Deep Learning for Retention Time Prediction in Reversed-Phase Liquid Chromatography. J. Chromatogr. A 2022, 1664, 462792. [Google Scholar] [CrossRef]
- Habra, H.; Meijer, J.L.; Shen, T.; Fiehn, O.; Gaul, D.A.; Fernández, F.M.; Rempfert, K.R.; Metz, T.O.; Peterson, K.E.; Evans, C.R.; et al. metabCombiner 2.0: Disparate Multi-Dataset Feature Alignment for LC-MS Metabolomics. Metabolites 2024, 14, 125. [Google Scholar] [CrossRef]
- Krokhin, O.V.; Spicer, V. Predicting Peptide Retention Times for Proteomics. Curr. Protoc. Bioinform. 2010, 13, 13–14. [Google Scholar] [CrossRef]
- Baczek, T.; Kaliszan, R. Predictions of Peptides’ Retention Times in Reversed-Phase Liquid Chromatography as a New Supportive Tool to Improve Protein Identification in Proteomics. Proteomics 2009, 9, 835–847. [Google Scholar] [CrossRef]
- Henneman, A.; Palmblad, M. Retention Time Prediction and Protein Identification. Methods Mol. Biol. 2020, 2051, 115–132. [Google Scholar] [CrossRef]
- Pfeifer, N.; Leinenbach, A.; Huber, C.G.; Kohlbacher, O. Statistical Learning of Peptide Retention Behavior in Chromatographic Separations: A New Kernel-Based Approach for Computational Proteomics. BMC Bioinform. 2007, 8, 468. [Google Scholar] [CrossRef] [PubMed]
- Kell, P.; Sidhu, R.; Qian, M.; Mishra, S.; Nicoli, E.-R.; D’Souza, P.; Tifft, C.J.; Gross, A.L.; Gray-Edwards, H.L.; Martin, D.R.; et al. A Pentasaccharide for Monitoring Pharmacodynamic Response to Gene Therapy in GM1 Gangliosidosis. eBioMedicine 2023, 92, 104627. [Google Scholar] [CrossRef]
- Field, A.P.; Gillett, R. How to Do a Meta-analysis. Brit. J. Math. Statis 2010, 63, 665–694. [Google Scholar] [CrossRef]
- O’Donnell, V.B.; Ekroos, K.; Liebisch, G.; Wakelam, M. Lipidomics: Current State of the Art in a Fast Moving Field. WIREs Mech. Dis. 2020, 12, e1466. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Sun, J.; Fan, S.; Yao, X.; Ran, X.; Zheng, C.; Huang, M.; Bi, H. Optimization of Lipid Extraction and Analytical Protocols for UHPLC-ESI-HRMS-Based Lipidomic Analysis of Adherent Mammalian Cancer Cells. Anal. Bioanal. Chem. 2017, 409, 5349–5358. [Google Scholar] [CrossRef]
- Cajka, T.; Smilowitz, J.T.; Fiehn, O. Validating Quantitative Untargeted Lipidomics Across Nine Liquid Chromatography–High-Resolution Mass Spectrometry Platforms. Anal. Chem. 2017, 89, 12360–12368. [Google Scholar] [CrossRef] [PubMed]
- Goracci, L.; Tortorella, S.; Tiberi, P.; Pellegrino, R.M.; Di Veroli, A.; Valeri, A.; Cruciani, G. Lipostar, a Comprehensive Platform-Neutral Cheminformatics Tool for Lipidomics. Anal. Chem. 2017, 89, 6257–6264. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A Lipidome Atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, E. Quantile Regression: A Short Story on How and Why. Stat. Model. 2018, 18, 203–218. [Google Scholar] [CrossRef]
- Koenker, R. Quantile Regression: 40 Years On. Annu. Rev. Econ. 2017, 9, 155–176. [Google Scholar] [CrossRef]
- Gottard, A.; Vannucci, G.; Grilli, L.; Rampichini, C. Mixed-Effect Models with Trees. Adv. Data Anal. Classif. 2023, 17, 431–461. [Google Scholar] [CrossRef]
- Alkharusi, H. Categorical Variables in Regression Analysis: A Comparison of Dummy and Effect Coding. Int. J. Educ. 2012, 4, 202. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Erion, G.G.; Lee, S.-I. Consistent Individualized Feature Attribution for Tree Ensembles. arXiv 2018, arXiv:1802.03888. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the Advances in Neural Information Processing Systems; Guyon, I., Luxburg, U.V., Bengio, S., Wallach, H., Fergus, R., Vishwanathan, S., Garnett, R., Eds.; Curran Associates, Inc.: Long Beach, CA, USA, 2017; Volume 30. [Google Scholar]
- Covert, I.; Lundberg, S.; Lee, S.-I. Feature Removal Is a Unifying Principle for Model Explanation Methods. arXiv 2020, arXiv:2011.03623. [Google Scholar] [CrossRef]
- Boyer, G. Chemparse 2022. Available online: https://pypi.org/project/chemparse/ (accessed on 25 October 2023).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Release Notes—SHAP Latest Documentation. Available online: https://shap.readthedocs.io/en/latest/release_notes.html (accessed on 25 October 2023).
- Da Costa-Luis, C.; Larroque, S.K.; Altendorf, K.; Mary, H.; Richardsheridan; Korobov, M.; Yorav-Raphael, N.; Ivanov, I.; Bargull, M.; Rodrigues, N.; et al. Tqdm: A Fast, Extensible Progress Bar for Python and CLI 2023. Available online: https://github.com/tqdm/tqdm (accessed on 25 October 2023).
- Raybaut, P. Spyder IDE. Available online: https://www.spyder-ide.org/ (accessed on 20 June 2023).
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P.A. Why Most Published Research Findings Are False. PLoS Med. 2005, 2, e124. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Jager, L.R. Is Most Published Research Really False? Annu. Rev. Stat. Appl. 2017, 4, 109–122. [Google Scholar] [CrossRef]
- Wood, P.L.; Cebak, J.E. Lipidomics Biomarker Studies: Errors, Limitations, and the Future. Biochem. Biophys. Res. Commun. 2018, 504, 569–575. [Google Scholar] [CrossRef]
- Onoja, A.; Von Gerichten, J.; Lewis, H.-M.; Bailey, M.J.; Skene, D.J.; Geifman, N.; Spick, M. Meta-Analysis of COVID-19 Metabolomics Identifies Variations in Robustness of Biomarkers. Int. J. Mol. Sci. 2023, 24, 14371. [Google Scholar] [CrossRef]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in Silico Tandem Mass Spectrometry Database for Lipid Identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef]
- Conroy, M.J.; Andrews, R.M.; Andrews, S.; Cockayne, L.; Dennis, E.A.; Fahy, E.; Gaud, C.; Griffiths, W.J.; Jukes, G.; Kolchin, M.; et al. LIPID MAPS: Update to Databases and Tools for the Lipidomics Community. Nucleic Acids Res. 2024, 52, D1677–D1682. [Google Scholar] [CrossRef] [PubMed]
- Ovčačíková, M.; Lísa, M.; Cífková, E.; Holčapek, M. Retention Behavior of Lipids in Reversed-Phase Ultrahigh-Performance Liquid Chromatography-Electrospray Ionization Mass Spectrometry. J. Chromatogr. A 2016, 1450, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Pchelkin, V.P. Calculations of the Hydrophobicity of Lipid Molecules by the Elution Strength of the Chromatographic Solvent. J. Anal. Chem. 2020, 75, 615–621. [Google Scholar] [CrossRef]
- White, J.B.; Trim, P.J.; Salagaras, T.; Long, A.; Psaltis, P.J.; Verjans, J.W.; Snel, M.F. Equivalent Carbon Number and Interclass Retention Time Conversion Enhance Lipid Identification in Untargeted Clinical Lipidomics. Anal. Chem. 2022, 94, 3476–3484. [Google Scholar] [CrossRef]
- Hughey, C.A.; Hendrickson, C.L.; Rodgers, R.P.; Marshall, A.G.; Qian, K. Kendrick Mass Defect Spectrum: A Compact Visual Analysis for Ultrahigh-Resolution Broadband Mass Spectra. Anal. Chem. 2001, 73, 4676–4681. [Google Scholar] [CrossRef]
- Naylor, B.C.; Catrow, J.L.; Maschek, J.A.; Cox, J.E. QSRR Automator: A Tool for Automating Retention Time Prediction in Lipidomics and Metabolomics. Metabolites 2020, 10, 237. [Google Scholar] [CrossRef]
- Vu, N.; Narvaez-Rivas, M.; Chen, G.-Y.; Rewers, M.J.; Zhang, Q. Accurate Mass and Retention Time Library of Serum Lipids for Type 1 Diabetes Research. Anal. Bioanal. Chem. 2019, 411, 5937–5949. [Google Scholar] [CrossRef] [PubMed]
- Rose, B.S.; May, J.C.; Picache, J.A.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Improving Confidence in Lipidomic Annotations by Incorporating Empirical Ion Mobility Regression Analysis and Chemical Class Prediction. Bioinformatics 2022, 38, 2872–2879. [Google Scholar] [CrossRef]
- Shimojima, M. Biosynthesis and Functions of the Plant Sulfolipid. Prog. Lipid Res. 2011, 50, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Abou-Elwafa Abdallah, M.; Nguyen, K.-H.; Ebele, A.J.; Atia, N.N.; Ali, H.R.H.; Harrad, S. A Single Run, Rapid Polarity Switching Method for Determination of 30 Pharmaceuticals and Personal Care Products in Waste Water Using Q-Exactive Orbitrap High Resolution Accurate Mass Spectrometry. J. Chromatogr. A 2019, 1588, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Ekroos, K.; McDonald, J.; Ahrends, R.; Liebisch, G.; Sandvig, K. Pitfalls in Lipid Mass Spectrometry of Mammalian Samples—A Brief Guide for Biologists. Nat. Rev. Mol. Cell Biol. 2024, 1471, 80. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS DIAL Identified Features | Actual tR (min) | Predicted tR (min) | Δ (min) |
|---|---|---|---|
| Inconsistencies in tR: saturation | |||
| TG C51H92O6 | 7.42 | 7.49 | 0.07 |
| TG C51H94O6 | 7.55 | 7.57 | 0.02 |
| TG C51H96O6 | 7.69 | 7.65 | −0.04 |
| TG C51H98O6 | 6.62 | 7.74 | 1.12 |
| Inconsistencies in tR: headgroups | |||
| DG C36H61D7O5 | 6.55 | 6.52 | −0.03 |
| PC C36H64NO8P | 6.50 | 5.42 | −1.08 |
| MS DIAL Identified Features | tR (min) | Lipostar Identified Features | tR (min) |
|---|---|---|---|
| Identification problems: co-elution of lipids | |||
| DG 34:0|DG 16:0_18:0 | 6.78 | DG (15:0/16:0/0:0) | 6.78 |
| TG 41:1;O|TG 9:0_17:0_15:1;O | 6.78 | DG (15:1/18:1/0:0) | 6.78 |
| Cer 42:2;O2|Cer 18:1;O2/24:1 | 6.80 | TG (13:0/13:0/16:0) | 6.78 |
| Cer 42:2;O2|Cer 18:1;O2/24:1 | 6.80 | DG (15:0/18:1/0:0) | 6.79 |
| Cer (51:1) | 6.80 | ||
| Identification problems: misidentifications | |||
| PC 37:7|PC 15:1_22:6 | 6.00 | PE (40:7) | 6.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Gerichten, J.; Saunders, K.; Bailey, M.J.; Gethings, L.A.; Onoja, A.; Geifman, N.; Spick, M. Challenges in Lipidomics Biomarker Identification: Avoiding the Pitfalls and Improving Reproducibility. Metabolites 2024, 14, 461. https://doi.org/10.3390/metabo14080461
von Gerichten J, Saunders K, Bailey MJ, Gethings LA, Onoja A, Geifman N, Spick M. Challenges in Lipidomics Biomarker Identification: Avoiding the Pitfalls and Improving Reproducibility. Metabolites. 2024; 14(8):461. https://doi.org/10.3390/metabo14080461
Chicago/Turabian Stylevon Gerichten, Johanna, Kyle Saunders, Melanie J. Bailey, Lee A. Gethings, Anthony Onoja, Nophar Geifman, and Matt Spick. 2024. "Challenges in Lipidomics Biomarker Identification: Avoiding the Pitfalls and Improving Reproducibility" Metabolites 14, no. 8: 461. https://doi.org/10.3390/metabo14080461