Abstract
Background/Objectives: During pregnancy, physiological changes in maternal circulating glucose levels and its metabolism are essential to meet maternal and fetal energy demands. Major changes in glucose metabolism occur throughout pregnancy and consist of higher insulin resistance and a compensatory increase in insulin secretion to maintain glucose homeostasis. For some women, this change is insufficient to maintain normoglycemia, leading to gestational diabetes mellitus (GDM), a condition characterized by maternal glucose intolerance and hyperglycaemia first diagnosed during the second or third trimester of pregnancy. GDM is diagnosed in approximately 14.0% of pregnancies globally, and it is often associated with short- and long-term adverse health outcomes in both mothers and offspring. Although recent studies have highlighted the role of genetic determinants in the development of GDM, research in this area is still lacking, hindering the development of prevention and treatment strategies. Methods: In this paper, we review recent advances in the understanding of genetic determinants of GDM and glycaemic traits during pregnancy. Results/Conclusions: Our review highlights the need for further collaborative efforts as well as larger and more diverse genotyped pregnancy cohorts to deepen our understanding of the genetic aetiology of GDM, address research gaps, and further improve diagnostic and treatment strategies.
1. Pathophysiology of GDM
In a healthy pregnancy, metabolic changes are essential to meet the energy expenditure required to support the growing fetus, with maternal glucose metabolism in particular providing necessary nutrition for healthy fetal growth. Changes in maternal glucose metabolism during pregnancy are characterized by elevated postprandial blood glucose levels, increased insulin resistance, and a compensatory increase in the secretion of insulin (Figure 1) [1,2,3,4,5,6,7,8,9,10,11,12]. These adaptations are essential to prepare the mother’s body for the metabolic demands of fetal growth, which includes transfer of nutrients from the mother to the developing fetus, as well as the provision of additional energy storage for both lactation and delivery [12].
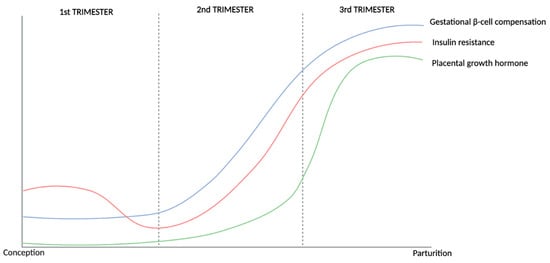
Figure 1.
Illustration depicting key changes in glucose metabolism that occur during a healthy pregnancy based on the literature reviewed and summarised above. Throughout pregnancy, the body undergoes a dynamic adjustment to maternal insulin resistance through β-cell compensation. This shift in insulin resistance is influenced by placental growth hormone, which acts locally to induce insulin resistance in maternal peripheral tissues. To counterbalance this, gestational β-cell compensation begins in the second trimester, marked by increased insulin secretion, and reaches its peak level in the third trimester, ensuring adequate glucose regulation despite increased insulin resistance. GDM, however, might occur if glucose utilization and the compensatory increase in insulin secretion are not sufficient to reduce and maintain blood glucose levels within the regulated range. GDM: gestational diabetes mellitus.
Fasting glucose (FG) levels decrease in the first trimester of pregnancy, likely as a result of increased plasma volume induced by early hormone changes, with these values stabilizing in the second trimester largely due to fetal utilization, before further decreasing during the third trimester [11,13]. An increased resistance to the action of insulin (compared with pre-pregnancy levels) is also observed in healthy pregnancies, resulting in higher postprandial glucose levels during gestation [1,6,7,11,14,15,16]. This insulin resistance increases as the pregnancy progresses through the second and third trimesters due to the influence of placental hormones (prolactin, human placental lactogen, and human placental growth hormone) and pro-inflammatory cytokines [1,2,3,4,5,11,17,18,19,20,21,22,23,24]. Additionally, this state of insulin resistance enhances endogenous glucose production and breakdown of stored fat, which consequently leads to a further increase in blood glucose and free fatty acid levels [23].
Although insulin resistance is a common and essential physiological change during pregnancy, gestational diabetes mellitus (GDM) manifests when the concomitant compensatory increase in insulin secretion that occurs during pregnancy does not counterbalance insulin resistance and, consequently, is unable to maintain glucose homeostasis [5]. This mechanism, known as β-cell compensation, is characterized by β-cell mass expansion and other key molecular changes that are necessary to increase insulin secretion and maintain normoglycemia [24,25,26,27,28,29,30]. As such, the inability of β-cells to produce more insulin can lead to maternal hyperglycaemia and increased levels of glucose crossing the placenta, causing the fetus to produce excess insulin, a known fetal growth factor, which can lead to excessive fetal growth and associated perinatal complications [30,31,32,33,34].
2. GDM Diagnosis and Screening
For GDM diagnosis, blood glucose levels during pregnancy are commonly measured as overnight FG as well as one- and two-hour (and/or more rarely three-hour) glucose plasma values post oral glucose tolerance test (OGTT). This test is typically conducted at 24–28 weeks of gestation, a period historically observed to show the greatest variation in glucose levels [35]. For pregnant individuals undergoing an OGTT, blood samples are first taken after an overnight fast to measure fasting glucose levels. Then, additional blood samples are collected at one, two, and/or three hours after the ingestion of a 75 g glucose solution to further assess glucose levels. Although all these measurements can be used to diagnose hyperglycaemia, higher FG levels are observed in response to insulin resistance whilst post-load OGTT plasma glucose values have been reported to better reflect glucose uptake in skeletal muscles and disturbances in insulin production and secretion (indicative of β-cell function), respectively [36,37,38].
The screening strategies and diagnostic criteria employed to identify GDM cases vary substantially across regions. These differences include variations in gestational age at screening, whether screening is targeted solely at high-risk women or universally applied as well as plasma cut-off levels used during the OGTT (for an overview of the variations in GDM diagnostic criteria, see Table 1). Despite ongoing research and discussions within the medical community, a consensus has yet to be reached regarding the optimal diagnostic and screening criteria for identifying mothers with GDM. As can be seen in Table 1, not only GDM screening approaches but also GDM diagnostic criteria vary considerably across regions and have changed over time. The most recent international criteria, developed by the International Association of Diabetes in Pregnancy Study Group (IADPSG) in 2010 and also supported by the World Health Organization (WHO), recommend a universal 75 g OGTT screening between 24 and 28 weeks of gestation. This recommendation is based on the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study findings, comprised of 23,316 women of multiple ethnic and geographic origins, which reported clear linear associations between maternal plasma glucose levels after a 75 g OGTT and a variety of maternal and fetal adverse outcomes such as large for gestational age, neonatal hypoglycaemia, and frequency of Caesarean sections [39,40,41,42]. The IADPSG committee suggested that prespecified odds of adverse outcomes should be used to set the thresholds of glucose levels for defining GDM cases based on the average glucose values at which odds for birth weight > 90th percentile, cord C-peptide > 90th percentile, and percent body fat > 90th percentile reached 1.75 times the estimated odds of these outcomes at mean glucose values (values based on adjusted logistic regression models) [39].

Table 1.
Summary of the different diagnostic criteria for gestational diabetes mellitus. OGTT: oral glucose tolerance test. FG: fasting glucose.
It is important to emphasize that although aiming to improve the diagnosis of GDM and find the best approach to reduce adverse outcomes (given existing resources and budgets), the thresholds set by IADPSG are arbitrary as there is no threshold effect (in the HAPO results). Consequently, this has led to criticism with regards to the implementation of a universal GDM screening using the IADPSG criteria as this approach could lead to the misdiagnosis of women with moderate hyperglycaemia and unnecessary prescription of medications in what was previously considered healthy pregnancies. Although seen to reduce the risk of offspring large for gestational age, preterm birth, and neonatal hypoglycaemia overall, this lower diagnostic threshold has also been shown to provide limited benefits to women without additional risk factors [43,44].
3. Epidemiology of GDM
GDM presents a significant challenge to maternal health, being diagnosed in approximately 14.0% of all pregnancies or roughly one in six births worldwide [45], though these figures are importantly influenced by marked variation in screening (i.e., universal vs. risk-based) and the thresholds used to diagnose GDM. Globally, the reported prevalence of GDM varies widely, with Middle Eastern and North African countries having the highest prevalence (27.6%) and North America and Caribbean regions having the lowest (7.1%) (Figure 2) [45]. Understanding the global burden of GDM has been challenging for decades due to the lack of uniform screening strategies and diagnostic criteria for GDM, variations in the prevalence of diagnosed type-I and II diabetes (as women with pre-existing diabetes follow a different obstetric care-path and are not screened or tested for GDM), and the diversity in antenatal health care practices across regions.
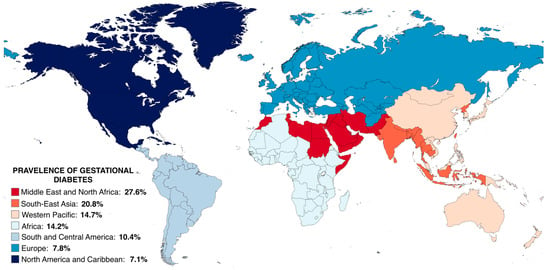
Figure 2.
Estimated prevalence of gestational diabetes across global regions; prevalences taken from Wang et al., 2022 [45] and visualised using MapChart. A fixed-effects meta-analysis of 57 studies covering 45 countries was performed, with the diagnostic criteria and universal OGTT strategy proposed by IADPSG, as well as the age group of 25–30 years, serving as benchmarks for standardizing the prevalence of GDM across various practices and age groups. The global standardized prevalence was reported to be 14.2%, with the pooled prevalence in pregnant women around 25–30 years of age being 27.6%, 20.8%, 14.7%, 14.2%, 10.4%, 7.8%, and 7.1% in the Middle East and North Africa (MENA), South-East Asia (SEA), Western Pacific (WP), Africa (AFR), South and Central America (SACA), Europe (EUR), and North America and Caribbean (NAC). Although the study controlled for diagnostic criteria, screening approach, and age group, population characteristics were not taken into account. Pooled prevalence should be interpreted with caution as it was calculated based on varying and arbitrary cut points. OGTT: Oral glucose tolerance test; IADPSG: International Association of the Diabetes and Pregnancy Study Group; GDM: gestational diabetes mellitus.
A systematic review and meta-analysis including up to 136,705 women was performed to compare the GDM prevalence after implementing the new IADPSG criteria with the GDM prevalence when older GDM criteria were used [46]. This study reported a 75% increase in the number of women diagnosed with GDM, with the overall effect estimates showing high heterogeneity in the pooled analysis. Subgroup analyses were undertaken for maternal age, BMI, study design, screening method, early screening, and use of modified IADPSG criteria, with a possible suggestion of such differences; however, there was no exploration of these differences statistically.
Whilst the adoption of a lower diagnostic threshold and recommendation of a universal screening have contributed to an increase in the number of identified GDM cases, an upward trend in GDM prevalence was already evident prior to the implementation of these new criteria [47,48,49,50,51,52]. Over the past few decades, for instance, the demographic profile of pregnant women has changed significantly, with women having children at a more advanced age in high- and some middle-income countries, and obesity rising globally, both of which are likely to have further contributed to an increase in the prevalence of GDM [47,53,54].
4. Aetiology of GDM
GDM has multiple genetic, lifestyle, and clinical risk factors contributing to disease onset and progression. However, these risk factors have been mostly investigated in traditional observational epidemiological studies, which are often prone to confounding by social, environmental, and behavioural factors. Genetics, however, provides an opportunity to inform on potential causal relationships between traditional risk factors and disease through the application of the genetic epidemiological technique “Mendelian randomization” (MR) [55]. In this method, genetic variants are used to proxy a traditional risk factor and estimate the causal relationship between the risk factor and the disease. Because genetic variants segregate independently of environmental confounders, the rationale is that they and the causal estimates derived from them should be less affected by confounding and other potential biases than traditional observational epidemiological studies. For example, MR studies of type-II diabetes (T2DM) have suggested that higher BMI and central fat distribution are key causes of T2DM, and that triglyceride-lowering drugs, as well as population-level interventions to reduce obesity, could help prevent T2DM [56,57,58]. Adequately powered two-sample MR studies are becoming increasingly possible as the size of GDM genome-wide association studies (GWASs) increases, enabling the investigation of causal factors underlying the risk of GDM. For example, obesity/overweight and high body mass index (BMI), conditions that are associated with both insulin resistance and inflammation [59,60,61,62,63,64,65,66,67,68], have been examined via MR. Overall, the evidence from these studies (and other designs involving, e.g., multivariable regression, paternal negative control, etc.) is robust, supporting the notion that a higher maternal BMI is causal for increased risk of GDM [69,70].
Another well-known risk factor for GDM is advanced maternal age at childbirth. Studies, including one with nearly a million participants, have consistently reported a progressive increase in GDM risk for mothers aged 25 years and older [45,47,71,72,73]. GDM recurrence has also been observed in nearly half of the women previously diagnosed with GDM in a Chinese cohort study (N = 10,151), with a recent and multi-ancestral meta-analysis, (N = 19,053) further suggesting that multiparous women have a higher recurrence rate of GDM compared to primiparous women (73% vs. 40%), although other factors like age and obesity might influence this relationship [47,74,75,76]. Additionally, women with a family history of diabetes are at increased risk of developing GDM, with a systematic review and meta-analysis (including 2697 women with a family history of diabetes mellitus and 29,134 women without) reporting up to 3.46 increased odds of developing GDM (95% CI: 2.80–4.27) compared to those who do not [77,78,79].
GDM also varies considerably across ethnicities [45,47,80,81]. For instance, a large, multiethnic, population-based study (N = 956,738) reported that South Asian women had 4.33 higher odds of developing GDM relative to Australian women [47]. Similarly, a study in the United States (N = 123,040) also reported a higher prevalence of GDM in Filipina and Asian women (10.9 and 10.2%, respectively), along with an intermediate prevalence among Hispanics (6.8%) and a lower prevalence in white Europeans and African American mothers (4.5 and 4.4%, respectively) [80]. Although this study had a large sample size, they lacked information on important risk factors, such as weight and family history of diabetes, which could influence the observed differences in prevalence across groups. Importantly, the robustness of findings for maternal age and ethnicity across many diverse studies supports them being true risk factors, although MR and other causal methods are difficult to implement for such characteristics.
5. Genetic Aetiology of GDM and Glycaemic Traits during Pregnancy
In addition to maternal lifestyle and environmental factors, genetics plays a role in GDM susceptibility. GWASs test for statistically robust associations between genetic variants and the trait of interest. In our review, we show how recent large-scale GWASs have provided estimates of the degree to which genetic variation influences liability to GDM and new insights into GDM disease aetiology; as mentioned previously, identified potential targets for pharmacotherapy; and, using the principles of MR, informed on potential public health preventive interventions [55,82,83].
5.1. Variation in Glycaemic Traits Explained by Genetics
Genetic variation is a ubiquitous contributor to individual differences in common complex traits and diseases. However, the genetic variants identified through large-scale GWASs that are robustly associated with glycaemic traits currently only explain a small proportion of the overall variance in these phenotypes. For instance, in the general population (i.e., outside of pregnancy), known genetic variants explain about 1.7% of the variance in HbA1c, 3% of FG variance, and between 4% and 14% (depending on ancestry) of the variance in 2-h glucose levels [84,85,86].
GWASs also permit the estimation of “SNP heritability” for common complex traits and diseases [87,88]. SNP heritability represents the proportion of trait variance that is tagged by SNPs on a microarray and consequently captures the sum total contribution of genome-wide significant variants and genetic variants of smaller effect scattered across the genome to overall trait heritability. It also represents that fraction of the phenotypic variance that is amenable to genetic discovery with increasing GWASs’ sample size. Recently, the SNP heritability for each of the glycaemic traits during pregnancy was estimated, with a study on East Asian women reporting estimates of approximately 5.3% for FG at weeks 16–18, 9.6% for FG at weeks 24–28, 10.2% for 1-h, and 7.8% for 2-h glucose post-OGTT at week 24–28 [83]. Interestingly, polygenic risk scores for FG during gestational weeks 24–32 in European mothers were previously reported to explain 4–7% of the variation in this trait, with studies further showing that the same FG-associated variants explained a similar proportion of variance both during and outside pregnancy [84,89,90]. Although it is interesting to see this concordance, it is also important to remember that all of these quantities (i.e., the proportion of phenotypic variance explained by a polygenic risk score, SNP heritability, and overall heritability) are population-specific ratio measures, which include environmental as well as genetic variation in the denominator and, thus, are expected to vary across different populations and circumstances/environments.
5.2. Robustly Associated Genetic Variants
In order to robustly identify common variants of small to moderate effect underlying common complex diseases with adequate statistical power, genome-wide association studies need to involve at least 2,000 cases and controls [91]. To the best of our knowledge, only six GWASs of GDM have been published to date [70,92,93,94,95,96], with the first study to robustly detect genome-wide significant loci for GDM being a Korean study (cases = 1,399; controls = 2,005) that identified genetic variants at the CDKAL1 and MTNR1B loci that were significantly associated with risk of GDM [95].
More recently, another study attempted to gain novel insights into the genetic architecture of GDM and address limitations associated with sample size by performing a multi-ancestry GWAS meta-analysis, which included 5,485 GDM cases and 347,856 healthy controls from various population groups, including Europeans, East Asians, South Asians, Hispanics/Latinos, and Africans [70]. This research effort, led by the GENetics of Diabetes In Pregnancy (GenDIP) Consortium, identified five loci at genome-wide levels of significance—three of them novel and two of them being the known CDKAL1 and MTNR1B loci [70]. Heterogeneity in estimated effect sizes across ancestries was present at two loci, CDKAL1 and CDKN2A-CDKN2B, potentially reflecting differences in linkage disequilibrium (LD) patterns between ancestries (although this might also indicate that the pathophysiological mechanisms driving glycaemic dysregulation in pregnancy may vary between ancestries, which needs to be further investigated) and emphasizing the need of more studies on under-represented populations [70].
The largest GWAS of GDM to date was undertaken in FinnGen, a study that combined genetic data with electronic health record data to support GWASs and other genetic analyses for many health outcomes [97]. The FinnGen GWAS comprised 12,332 GDM cases (i.e., with a GDM diagnosis listed in their health record) and 131,109 parous female controls from Finland, along with an independent sample of 8,931 cases and 170,809 controls for replication (from both Finland and Estonia). The study identified thirteen genome-wide significant loci associated with GDM, with eight of the loci being novel [96]. Further analyses were also performed to determine the extent to which each locus was associated with T2DM and GDM based on their effect sizes for each condition. Loci showing GDM-predominant effects were mapped to genes linked to islet cells, central glucose homeostasis, steroidogenesis, and placental expression—these GDM-predominant effect loci include (GCKR, SPC25-G6PC2, PCSK1, ESR1, MTNR1B, NEDD1, CMIP, and MAP3K15), whereas the loci at CDKAL1, TCF7L2 and CCND2 involved T2DM predominant effects [96]. Nevertheless, the estimates of allelic effects at these loci were almost entirely in the same direction [96].
As it is possible to see in Table 2, several possible loci have been implicated in GDM [70,93,94,95,96]. However, it is important to note that while numerous genetic variants associated with GDM have been identified through GWASs, the causal genes for most loci remain unknown. Hence, this table lists candidate genes based on their proximity to the association signal, along with any additional evidence supporting their potential causality, with the caveat that these are not definitively proven causal genes but are considered candidates pending further investigation.
Table 2.
Overview of the candidate genes implicated by reported GWAS associations, describing their respective function and previously reported associations. SNP; Single-Nucleotide Polymorphism. * Associations reported in previous Genome-Wide Association Studies (GWASs) that reached the genome-wide significance threshold of 5 10−8.
As glucose measurements can reflect changes in glucose metabolism across specific timepoints, and GDM is diagnosed based on an arbitrary threshold applied to these underlying quantitative traits, investigating the genetics of fasting glucose and post-OGTT glucose levels can provide deeper insights into the genetic basis of GDM. Hence, the genetic determinants of fasting and postprandial blood glucose during pregnancy have been investigated in a few studies [89,92,202,204]. One of these studies investigated 4437 mothers of different ancestries and identified five loci associated with FG (i.e., GCKR, G6PC2, PCSK1, PPP1R3B, and MTNR1B) [202]. Additionally, an association between 1-h glucose post-OGTT and variants in MTNR1B as well as 2-h glucose post-OGTT and variants in HKDC1 has also been detected. While these associations showed important genetic determinants of glycaemic traits during pregnancy, subsequent studies have failed to replicate such associations due to limited sample sizes [89,94,204].
A recent study, however, explored the association between genetic variants and both GDM and several glycaemic traits (such as FG, 1-h post OGTT, and 2-h post OGTT glucose levels) in up to 26,751 East Asian mothers [92]. Although the sample size for GDM was significantly smaller than the ones from the two studies previously discussed (with solely 3317 cases and 19,565 controls), the sample sizes for the quantitative traits analysed were the largest to date, with FG measurements on 26,751 mothers at weeks 16–18 of gestation along with information on FG (N = 24,929), 1-h glucose post OGTT (N = 24,931), and 2-h glucose post OGTT (N = 24,931) values at gestational weeks 24–28. Overall, nine loci were associated with FG at gestational weeks 16–18, ten with FG at gestational weeks 24–28, seven with 1-h glucose post OGTT, and, finally, four genes associated with 2-h glucose levels post OGTT (both at gestational weeks 24–28). The genetic determinants between fasting (or baseline) glycaemic levels and glycaemic values after OGTT have been observed to be substantially different—although it is important to note that FG was measured during gestational weeks 16–18 and weeks 24–28 while 1-h and 2-h post-OGGT levels investigated were measured solely during weeks 24–28. For instance, ABCB11, GCK, LOC101929710, and FOXA2 were only detected in the baseline glycaemic level analyses but not after OGTT. Additionally, CDKAL1, while not associated with FG at weeks 16–18, was seen to be significant in FG, 1-h glucose, and 2-h glucose post OGTT analyses at weeks 24–28 and HKDC1, showing a strong association with FG and 2-h glucose values post OGTT at weeks 24–28 but not with other glycaemic measurements. Further, MTNR1B was detected in all analyses, although the strength of association differed across timepoints, showing a stronger relationship with 1-h and 2-h post OGTT glucose values.
Interestingly, many genetic associations with glycaemic traits during pregnancy have been detected in both non-pregnant individuals and in association with GDM (Figure 3 shows a summary of the shared and distinct genetic variants). For example, at the genome-wide level, two loci associated with 2-h glucose post-OGTT (HKDC1 and CDKAL1) and twelve loci associated with FG, including GCKR, G6PC2, PCSK1, MTNR1B, ABCB11, RFX6, CDKAL1, CAMK2B, KANK1, GCK, and FOXA2, have been observed in both pregnant and non-pregnant populations (although proper colocalization analyses are needed to assess whether the same variants are implicated during and outside of pregnancy as this approach uses GWASs data to identify shared genetic factors across multiple related traits, helping pinpoint causal genes and mechanisms in complex diseases [269]) [66,92,95,96,110,202]. Similar trends were seen when comparing East Asian mothers in a large study on glycaemic traits in the general population (comprising up to 281,416 individuals): ABCB11 and FOXA2 were found in FG analyses but not in 2-h post-OGTT analyses, and CDKAL1 was associated with both FG and 2-h glucose values [92,110]. Differences between studies included HKDC1 being linked to both FG and 2-h glucose in pregnant East Asian mothers but only to 2-h glucose in the general population, and MTNR1B being significant in all glycaemic trait analyses during pregnancy but only in FG analysis in the general population [92,110]. The study of glycaemic traits in the non-pregnant population, however, only investigated FG and 2-h post-OGTT glucose values, as it is not common practice to measure 1-h glucose values post-OGTT outside of pregnancy. Hence, it is still unclear whether the genetic determinants of 1-h glucose values during and outside of pregnancy differ [110]. Although the genetic architecture of glycaemic traits during and outside pregnancy has been suggested to be shared for the most part, the genetic determinants of 2-h post OGTT glucose in pregnant women was reported to differ from the ones in the non-pregnant population [89,90]. Overall, findings suggest that although FG levels remain relatively stable outside and during gestation, the postprandial glucose levels tend to differ in order to meet the metabolic requirements imposed by fetal growth [7,12].
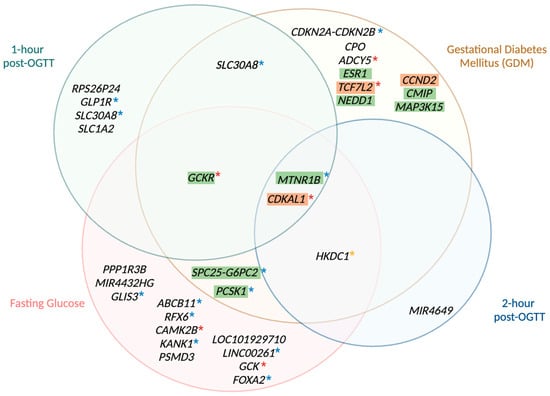
Figure 3.
Venn diagram of genetic loci associated with gestational diabetes, 1-h glucose, fasting glucose, and 2-h glucose. This Venn diagram depicts the genetic loci that harbour variants associated with GDM, fasting glucose levels, 1-h glucose levels post OGTT, and 2-h glucose levels post OGTT, as identified by multiple genome-wide association studies of pregnant women. Each circle represents the genetic variants linked to one of the four traits. The distinct, non-overlapping areas of each circle indicate genetic variants uniquely associated with each trait, while the overlapping regions illustrate genetic variants shared between two or more traits. The central overlapping area represents variants common to all four traits. All genetic variants included in this diagram reached genome-wide significance (p < 5 × 10−8) in the studies of Kwak et al. [95], Hayes et al. [202], Pervjakova et al. [70], Elliot et al. [96], and Zhen et al. [92]. Loci with GDM-predominant effects are highlighted in green, whereas loci with type-2 diabetes mellitus predominant effects are highlighted in orange as reported by Elliot et al. [96]. Loci not highlighted remain unclassified. Loci also detected by Chen et al. [110], in the general, non-pregnant population can be distinguished based on the asterisks (*), with blue asterisks indicating associations with fasting glucose values, yellow asterisks indicating associations with 2-h post-OGTT glucose values, and red asterisks indicating associations with both fasting and 2-h post-OGTT glucose values. Colocalization analyses, however, are needed to properly compare variants inside and outside of pregnancy [269]. GDM: gestational diabetes mellitus; OGTT: oral glucose tolerance test.
Despite recent advances in this field, the current body of research on genetic associations with GDM and glucose measurements during pregnancy remains limited, especially when compared to the extensive literature available on T2DM and glycaemic traits in the general population. Efforts to increase the genetic diversity in GWASs of GDM, especially the inclusion of underrepresented groups, are a key priority as genetic effects may vary between ancestries, a higher GDM prevalence is observed in some underrepresented groups, and the majority of the findings to date are based on European or East Asian populations [269]. Further, since identifying causal variants and the genes involved is a challenge due to the complex correlational structure of the genome, the inclusion of other ancestries where LD patterns may vary provides opportunities for improved fine mapping of genetic loci. Nevertheless, accurately addressing LD remains a challenge due to the inherent complexity of genomic structures and the need for comprehensive, high-resolution data across diverse populations.
5.3. Genetic Insights into the Relationship between GDM and T2DM
GWASs provide an opportunity to elucidate the degree to which diseases with different clinical presentations (e.g., T2DM and GDM) represent the same underlying disorder by examining the genetic similarity between them. This is typically performed on a locus-by-locus basis, as well as genome-wide, using methods like LD score regression, which estimates the overall genetic correlation between the traits [88]. The majority of the GWASs of GDM reported a substantial shared genetic aetiology between GDM and T2DM, with only a few genome-wide significant loci for GDM not being significantly associated with T2DM [66,92,93,94,95,96]. The largest GDM GWAS to date further used a new method called SCOUTJOY (Significant Cross-trait OUtliers and Trends in JOint York regression) to compare effect sizes of GDM-associated loci with those of T2DM. This approach evaluates if observed effect sizes across top hits conform to a uniform relationship, or whether some loci exhibit stronger associations with GDM or T2DM, while also accounting for sample overlap and estimation errors specific to each phenotype [96]. Overall, the authors reported significant heterogeneity in expected effect sizes across many of the loci, suggesting some genetic differences between the two conditions.
Additionally, the two largest GWASs of GDM reported a genetic correlation between GDM and T2DM of around 0.70, suggesting that whilst GDM and T2DM share much of their aetiology, they may also have distinct components that contribute to their individual genetic architectures [66,96]. However, it is worth bearing in mind that these genetic correlation estimates are based on relatively small numbers of GDM cases (i.e., cases = 5485, controls = 347,856 [66]; and cases = 12,332, controls = 131,109 [96]), which also include potentially less reliable self-reported diagnoses (e.g., in the UK Biobank). This contrasts with the very large sample sizes of recent T2DM GWASs, which involve up to 428,452 cases and 2,107,149 controls [140,270,271,272]. Overall, the relatively small sample size of GDM (compared to T2DM) and the degree to which the diagnoses of GDM can be trusted limit current attempts to understand the extent to which they represent the same condition (i.e., the physiological stress of pregnancy unmasks a predisposition to T2DM that becomes diagnosed as GDM), or whether there are distinct determinants of both.
6. Relationship between GDM and Short- and Long-Term Adverse Health Outcomes
GDM, like many other pregnancy complications, can negatively impact both maternal and fetal health. Although a variety of short- and long-term adverse outcomes are associated with GDM, evidence is mostly based on observational epidemiological studies, and so it is unclear whether these relationships represent causality or confounding through, e.g., shared genetics. To the best of our knowledge, no studies to date have attempted to examine potential causal relationships between GDM and either maternal or fetal long-term outcomes using Mendelian randomization [55]. This is partly a consequence of the limited number of genetic variants uniquely associated with GDM (i.e., as opposed to variants associated with both GDM and T2DM) and highlights the need for increasingly large GWASs to detect such variants that could then be used in MR analyses. As such, in this section, we briefly discuss the observational association between GDM and some adverse outcomes with the caveat that these associations require validation using causal inference methods.
In the short-term, GDM (similar to T2DM and type-1 diabetes), is associated with adverse obstetric and neonatal outcomes [273,274,275,276,277,278,279,280,281,282,283,284]. A multi-ancestry meta-analysis of 14,033,990 pregnancies highlighted the increased risk of hypertensive disorders of pregnancy, induction of labour, caesarean delivery, offspring large-for-gestational-age, preterm birth, and admission to the neonatal intensive care unit in mothers with GDM [285]. Although this study was well-powered, there was substantial heterogeneity in the magnitude of association across studies, likely reflecting different methods of GDM screening/diagnosis, diverse population demographics, and methodological variations [285]. These same adverse outcomes, however, were also reported by a recent meta-analysis of 7,506,061 pregnancies, with GDM being further associated with increased odds of low one-minute Apgar score, macrosomia, respiratory distress syndrome, and neonatal jaundice [286]. Further supporting these findings, a separate study demonstrated a consistent graded linear association between both maternal fasting and post-OGTT glucose concentration and clinically relevant perinatal outcomes—such as caesarean section, induction of labour, large for gestational age, macrosomia, and shoulder dystocia—with no clear evidence of a threshold effect, a trend also seen in the HAPO study [279,287]. Interestingly, recent research has also emphasized the critical role of glucose in fetal growth, with various maternal and fetal proteins being involved in glucose homeostasis and energy metabolism and having a potential effect on offspring birth weight [288].
As for the long-term impacts, the majority of the studies have focused on later-life outcomes, often chronic conditions, displayed by mothers who have had a GDM diagnosis. For example, it is well-established that GDM is associated with an increased risk of T2DM, with studies showing that a previous diagnosis of GDM may carry an 8–10-fold higher risk of T2DM, and the cumulative incidence can increase markedly in the first five years after delivery [289,290,291,292,293,294,295,296]. However, as discussed previously, the discrepancy between studies on GDM and T2DM further limits the important exploration of whether they are the same or distinct conditions. Apart from T2DM, GDM has also been reported to be associated with a variety of other maternal chronic conditions, including cardiovascular diseases (CVDs) and metabolic syndrome [297,298,299,300], although causal evidence is lacking. For instance, two large meta-analyses (one including 5,390,591 women and the other 3,417,020) reported that women diagnosed with GDM may have a 2-fold higher risk of future cardiovascular events, with a 2.3-fold increased risk being observed in the first decade postpartum [297,298]. In addition, a meta-analysis containing up to 5832 women also reported that mothers with a history of GDM may have up to 4-fold increased odds of developing metabolic syndrome compared to those without [299]. Another meta-analysis (N = 13,390 participants) corroborated these findings and further reported that women may be diagnosed with metabolic syndrome as early as one year postpartum [300]. The meta-analyses of CVD and metabolic syndrome, however, all presented significant heterogeneity potentially due to varying follow-up durations; geographic biases; inconsistent definitions of GDM, CVD, and metabolic syndrome; age-related effects; potential inclusion of women with pre-existing diabetes; and a small number of studies, which together hinder precise estimation of the true risk of GDM for both CVD and metabolic syndrome [297,298,299,300].
Although the long-term impacts of GDM on offspring health have also been investigated, research in this area continues to lack long-term follow-up into adulthood, for which large trans-generational cohorts are required as well as causal analysis. The largest meta-analyses have reported that GDM (or diabetes during pregnancy in general) is associated with an increase in the child’s risk of metabolic syndrome (N = 4421) and leads to higher offspring systolic blood pressure (N = 62,344), blood glucose (N = 6423), and BMI (N = 27,311), with these associations with BMI and systolic blood pressure also being observed in a recent meta-analysis with up to 8759 participants [300,301,302,303]. In addition, studies have reported a possible association with long-term hospitalizations with diagnoses of endocrine morbidity such as diabetes mellitus and obesity in the offspring (N of the retrospective cohort study = 231,271), as well as increased odds of childhood obesity (N of the cross-sectional study = 4740)—although association with obesity was no longer significant after adjusting for maternal BMI [304,305]. Impaired glucose tolerance and future risk of T2DM has also been reported; however, the studies were extremely underpowered with sample sizes of 255 and 597, respectively [306,307]. The meta-analyses and epidemiological studies discussed also contain limitations related to variations in phenotypic measurement (e.g., the age at which offspring blood pressure was measured varied), differences in GDM definition, and inability to properly control for confounders often due to lack of information on those factors. Causal evidence is limited, and it is important to emphasize that studies on long-term health outcomes in offspring of mothers with GDM have not taken the correlation between maternal and offspring genetics into account, and these associations could be due to genetic pleiotropy [308,309,310,311,312,313].
7. Conclusions
GDM is a significant global health challenge, being diagnosed in approximately 14.0% of pregnancies worldwide. Its prevalence varies widely across regions and is influenced by factors such as maternal age, ancestry, obesity, and family history of diabetes. Early diagnosis and management are crucial to mitigating the adverse outcomes associated with GDM for both mothers and their offspring. However, challenges persist in understanding the underlying biological mechanisms underpinning this condition, hampering efforts to identify affected women and further improve treatment strategies. Despite recent advances and the use of GWASs to understand the genetic landscape of GDM and glycaemic traits during pregnancy, further research is needed to fully elucidate the genetic factors contributing to GDM onset and recurrence, as well as the degree to which these factors are distinct from T2DM.
8. Future Directions
Despite recent advances, the current body of research on genetic associations with GDM and glucose measurements during pregnancy remains limited, especially when compared to the extensive research on T2DM and glycaemic traits in the general population. To further understand the genetic influences on glucose metabolism during pregnancy, larger pregnancy cohorts and international collaborative efforts are required, with a major focus on increasing the genetic diversity within studies. Additionally, there is a growing potential for drug target MR studies as seen in the context of T2DM, which could identify medications for better treatment of GDM and test their safety, particularly as more proteomic data become available in pregnancy. By harnessing the power of large-scale studies, current limitations can be addressed, improving our understanding of GDM pathogenesis and facilitating the development of more effective diagnostic and therapeutic strategies to improve maternal and fetal outcomes.
Author Contributions
Conceptualization, C.B.N. and G.-H.M.; writing—original draft preparation, C.B.N. and G.-H.M.; writing—review and editing, C.B.N., G.-H.M., M.C.B., R.M.F., D.A.L., E.Q. and D.M.E.; supervision, G.-H.M. and D.M.E. All authors have read and agreed to the published version of the manuscript.
Funding
G.-H.M. is the recipient of an Australian Research Council Discovery Early Career Award (Project number: DE220101226) funded by the Australian Government and supported by the Research Council of Norway (Project grant: 325640). D.M.E. is supported by a National Health and Medical Research Council Investigator Award (2017942). R.M.F. is supported by a Wellcome Senior Research Fellowship (WT220390) and a grant from the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number R01HD101669. D.A.L. and M.C.B. work in a unit that is supported by the University of Bristol and the UK Medical Research Council (MC_UU_00032/05), and D.A.L.’s contribution was supported by the British Heart Foundation (CH/F/20/90003) (£508,351. 2021–2026). This research was funded in part by the Wellcome Trust (Grant number: WT220390). For the purpose of Open Access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Catalano, P.M.; Roman-Drago, N.M.; Amini, S.B.; Sims, E.A. Longitudinal Changes in Body Composition and Energy Balance in Lean Women with Normal and Abnormal Glucose Tolerance during Pregnancy. Am. J. Obstet. Gynecol. 1998, 179, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, R.L.; Brelje, T.C. Adaptation of Islets of Langerhans to Pregnancy: Beta-Cell Growth, Enhanced Insulin Secretion and the Role of Lactogenic Hormones. Horm. Metab. Res. 1997, 29, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Bonner-Weir, S.; Deery, D.; Leahy, J.L.; Weir, G.C. Compensatory Growth of Pancreatic Beta-Cells in Adult Rats after Short-Term Glucose Infusion. Diabetes 1989, 38, 49–53. [Google Scholar] [CrossRef]
- Montaña, E.; Bonner-Weir, S.; Weir, G.C. Transplanted Beta Cell Response to Increased Metabolic Demand. Changes in Beta Cell Replication and Mass. J. Clin. Investig. 1994, 93, 1577–1582. [Google Scholar] [CrossRef]
- Buchanan, T.A.; Metzger, B.E.; Freinkel, N.; Bergman, R.N. Insulin Sensitivity and B-Cell Responsiveness to Glucose during Late Pregnancy in Lean and Moderately Obese Women with Normal Glucose Tolerance or Mild Gestational Diabetes. Am. J. Obstet. Gynecol. 1990, 162, 1008–1014. [Google Scholar] [CrossRef]
- Catalano, P.M.; Tyzbir, E.D.; Wolfe, R.R.; Calles, J.; Roman, N.M.; Amini, S.B.; Sims, E.A. Carbohydrate Metabolism during Pregnancy in Control Subjects and Women with Gestational Diabetes. Am. J. Physiol. 1993, 264 Pt 1, E60–E67. [Google Scholar] [CrossRef]
- Frøslie, K.F.; Røislien, J.; Qvigstad, E.; Godang, K.; Bollerslev, J.; Henriksen, T.; Veierød, M.B. Shape Information in Repeated Glucose Curves during Pregnancy Provided Significant Physiological Information for Neonatal Outcomes. PLoS ONE 2014, 9, e90798. [Google Scholar] [CrossRef] [PubMed]
- Lesser, K.B.; Carpenter, M.W. Metabolic Changes Associated with Normal Pregnancy and Pregnancy Complicated by Diabetes Mellitus. Semin. Perinatol. 1994, 18, 399–406. [Google Scholar] [PubMed]
- Metzger, B.E. Biphasic Effects of Maternal Metabolism on Fetal Growth. Quintessential Expression of Fuel-Mediated Teratogenesis. Diabetes 1991, 40 (Suppl. S2), 99–105. [Google Scholar] [CrossRef]
- Metzger, B.E.; Phelps, R.L.; Freinkel, N.; Navickas, I.A. Effects of Gestational Diabetes on Diurnal Profiles of Plasma Glucose, Lipids, and Individual Amino Acids. Diabetes Care 1980, 3, 402–409. [Google Scholar] [CrossRef]
- Di Cianni, G.; Miccoli, R.; Volpe, L.; Lencioni, C.; Del Prato, S. Intermediate Metabolism in Normal Pregnancy and in Gestational Diabetes. Diabetes Metab. Res. Rev. 2003, 19, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Hadden, D.R.; McLaughlin, C. Normal and Abnormal Maternal Metabolism during Pregnancy. Semin. Fetal. Neonatal Med. 2009, 14, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Tyzbir, E.D.; Roman, N.M.; Amini, S.B.; Sims, E.A. Longitudinal Changes in Insulin Release and Insulin Resistance in Nonobese Pregnant Women. Am. J. Obstet. Gynecol. 1991, 165 Pt 1, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Tyzbir, E.D.; Wolfe, R.R.; Roman, N.M.; Amini, S.B.; Sims, E.A. Longitudinal Changes in Basal Hepatic Glucose Production and Suppression during Insulin Infusion in Normal Pregnant Women. Am. J. Obstet. Gynecol. 1992, 167 Pt 1, 913–919. [Google Scholar] [CrossRef]
- Butte, N.F. Carbohydrate and Lipid Metabolism in Pregnancy: Normal Compared with Gestational Diabetes Mellitus12345. Am. J. Clin. Nutr. 2000, 71, 1256S–1261S. [Google Scholar] [CrossRef]
- Sonagra, A.D.; Biradar, S.M.; Dattatreya K.; Murthy D.S., J. Normal Pregnancy—A State of Insulin Resistance. J. Clin. Diagn. Res. 2014, 8, CC01–CC03. [Google Scholar] [CrossRef]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef]
- Buchanan, T.A.; Xiang, A.; Kjos, S.L.; Watanabe, R. What Is Gestational Diabetes? Diabetes Care 2007, 30 (Suppl. S2), S105–S111. [Google Scholar] [CrossRef]
- Barbour, L.A.; McCurdy, C.E.; Hernandez, T.L.; Kirwan, J.P.; Catalano, P.M.; Friedman, J.E. Cellular Mechanisms for Insulin Resistance in Normal Pregnancy and Gestational Diabetes. Diabetes Care 2007, 30 (Suppl. S2), S112–S119. [Google Scholar] [CrossRef] [PubMed]
- Vrachnis, N.; Belitsos, P.; Sifakis, S.; Dafopoulos, K.; Siristatidis, C.; Pappa, K.I.; Iliodromiti, Z. Role of Adipokines and Other Inflammatory Mediators in Gestational Diabetes Mellitus and Previous Gestational Diabetes Mellitus. Int. J. Endocrinol. 2012, 2012, 549748. [Google Scholar] [CrossRef]
- Kirwan, J.P.; Hauguel-De Mouzon, S.; Lepercq, J.; Challier, J.-C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. TNF-Alpha Is a Predictor of Insulin Resistance in Human Pregnancy. Diabetes 2002, 51, 2207–2213. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Vilariño-García, T.; Guadix, P.; Dueñas, J.L.; Sánchez-Margalet, V. Leptin and Nutrition in Gestational Diabetes. Nutrients 2020, 12, 1970. [Google Scholar] [CrossRef]
- Phelps, R.L.; Metzger, B.E.; Freinkel, N. Carbohydrate Metabolism in Pregnancy: XVII. Diurnal Profiles of Plasma Glucose, Insulin, Free Fatty Acids, Triglycerides, Cholesterol, and Individual Amino Acids in Late Normal Pregnancy. Am. J. Obstet. Gynecol. 1981, 140, 730–736. [Google Scholar] [CrossRef]
- Ernst, S.; Demirci, C.; Valle, S.; Velazquez-Garcia, S.; Garcia-Ocaña, A. Mechanisms in the Adaptation of Maternal β-Cells during Pregnancy. Diabetes Manag. Lond. Engl. 2011, 1, 239–248. [Google Scholar] [CrossRef]
- Lain, K.Y.; Catalano, P.M. Metabolic Changes in Pregnancy. Clin. Obstet. Gynecol. 2007, 50, 938–948. [Google Scholar] [CrossRef]
- Butler, A.E.; Cao-Minh, L.; Galasso, R.; Rizza, R.A.; Corradin, A.; Cobelli, C.; Butler, P.C. Adaptive Changes in Pancreatic Beta Cell Fractional Area and Beta Cell Turnover in Human Pregnancy. Diabetologia 2010, 53, 2167–2176. [Google Scholar] [CrossRef] [PubMed]
- Rieck, S.; Kaestner, K.H. Expansion of Beta-Cell Mass in Response to Pregnancy. Trends Endocrinol. Metab. TEM 2010, 21, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Toyofuku, Y.; Lynn, F.C.; Chak, E.; Uchida, T.; Mizukami, H.; Fujitani, Y.; Kawamori, R.; Miyatsuka, T.; Kosaka, Y.; et al. Serotonin Regulates Pancreatic Beta Cell Mass during Pregnancy. Nat. Med. 2010, 16, 804–808. [Google Scholar] [CrossRef]
- Karnik, S.K.; Chen, H.; McLean, G.W.; Heit, J.J.; Gu, X.; Zhang, A.Y.; Fontaine, M.; Yen, M.H.; Kim, S.K. Menin Controls Growth of Pancreatic Beta-Cells in Pregnant Mice and Promotes Gestational Diabetes Mellitus. Science 2007, 318, 806–809. [Google Scholar] [CrossRef]
- Ryan, E.A.; Enns, L. Role of Gestational Hormones in the Induction of Insulin Resistance. J. Clin. Endocrinol. Metab. 1988, 67, 341–347. [Google Scholar] [CrossRef]
- Desoye, G.; Nolan, C.J. The Fetal Glucose Steal: An Underappreciated Phenomenon in Diabetic Pregnancy. Diabetologia 2016, 59, 1089–1094. [Google Scholar] [CrossRef]
- Lindsay, R.S.; Walker, J.D.; Halsall, I.; Hales, C.N.; Calder, A.A.; Hamilton, B.A.; Johnstone, F.D.; Scottish Multicentre Study of Diabetes in Pregnancy. Insulin and Insulin Propeptides at Birth in Offspring of Diabetic Mothers. J. Clin. Endocrinol. Metab. 2003, 88, 1664–1671. [Google Scholar] [CrossRef][Green Version]
- Silverman, B.L.; Landsberg, L.; Metzger, B.E. Fetal Hyperinsulinism in Offspring of Diabetic Mothers. Association with the Subsequent Development of Childhood Obesity. Ann. N. Y. Acad. Sci. 1993, 699, 36–45. [Google Scholar] [CrossRef]
- Dornhorst, A.; Nicholls, J.S.; Ali, K.; Andres, C.; Adamson, D.L.; Kelly, L.F.; Niththyananthan, R.; Beard, R.W.; Gray, I.P. Fetal Proinsulin and Birth Weight. Diabet. Med. 1994, 11, 177–181. [Google Scholar] [CrossRef]
- Lawlor, D.A. The Society for Social Medicine John Pemberton Lecture 2011. Developmental Overnutrition—An Old Hypothesis with New Importance? Int. J. Epidemiol. 2013, 42, 7–29. [Google Scholar] [CrossRef]
- McAuley, K.A.; Williams, S.M.; Mann, J.I.; Walker, R.J.; Lewis-Barned, N.J.; Temple, L.A.; Duncan, A.W. Diagnosing Insulin Resistance in the General Population. Diabetes Care 2001, 24, 460–464. [Google Scholar] [CrossRef]
- Joshipura, K.J.; Andriankaja, M.O.; Hu, F.B.; Ritchie, C.S. Relative Utility of 1-Hr Oral Glucose Tolerance Test as a Measure of Abnormal Glucose Homeostasis. Diabetes Res. Clin. Pract. 2011, 93, 268–275. [Google Scholar] [CrossRef]
- Lee, S.; Choi, S.; Kim, H.J.; Chung, Y.-S.; Lee, K.W.; Lee, H.C.; Huh, K.B.; Kim, D.J. Cutoff Values of Surrogate Measures of Insulin Resistance for Metabolic Syndrome in Korean Non-Diabetic Adults. J. Korean Med. Sci. 2006, 21, 695–700. [Google Scholar] [CrossRef]
- International Association of Diabetes and Pregnancy Study Groups Consensus Panel; Metzger, B.E.; Gabbe, S.G.; Persson, B.; Buchanan, T.A.; Catalano, P.A.; Damm, P.; Dyer, A.R.; Leiva, A.d.; Hod, M. International Association of Diabetes and Pregnancy Study Groups Recommendations on the Diagnosis and Classification of Hyperglycemia in Pregnancy. Diabetes Care 2010, 33, 676–682. [Google Scholar] [CrossRef]
- HAPO Study Cooperative Research Group. The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. Int. J. Gynecol. Obstet. 2002, 78, 69–77. [Google Scholar] [CrossRef]
- The HAPO Study Cooperative Research Group. Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study: Associations With Neonatal Anthropometrics. Diabetes 2009, 58, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; McIntyre, H.D.; et al. Hyperglycemia and Adverse Pregnancy Outcomes. Obstet. Anesth. Dig. 2009, 29, 39. [Google Scholar] [CrossRef]
- Aubry, E.M.; Raio, L.; Oelhafen, S. Effect of the IADPSG Screening Strategy for Gestational Diabetes on Perinatal Outcomes in Switzerland. Diabetes Res. Clin. Pract. 2021, 175, 108830. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.S.; Sletner, L.; Jenum, A.K.; Øverby, N.C.; Stafne, S.N.; Qvigstad, E.; Pripp, A.H.; Sagedal, L.R. Employing Fasting Plasma Glucose to Safely Limit the Use of Oral Glucose Tolerance Tests in Pregnancy: A Pooled Analysis of Four Norwegian Studies. Front. Endocrinol. 2023, 14, 1278523. [Google Scholar] [CrossRef]
- Wang, H.; Li, N.; Chivese, T.; Werfalli, M.; Sun, H.; Yuen, L.; Hoegfeldt, C.A.; Elise Powe, C.; Immanuel, J.; Karuranga, S.; et al. IDF Diabetes Atlas: Estimation of Global and Regional Gestational Diabetes Mellitus Prevalence for 2021 by International Association of Diabetes in Pregnancy Study Group’s Criteria. Diabetes Res. Clin. Pract. 2022, 183, 109050. [Google Scholar] [CrossRef]
- Saeedi, M.; Cao, Y.; Fadl, H.; Gustafson, H.; Simmons, D. Increasing Prevalence of Gestational Diabetes Mellitus When Implementing the IADPSG Criteria: A Systematic Review and Meta-Analysis. Diabetes Res. Clin. Pract. 2021, 172, 108642. [Google Scholar] [CrossRef]
- Anna, V.; van der Ploeg, H.P.; Cheung, N.W.; Huxley, R.R.; Bauman, A.E. Sociodemographic Correlates of the Increasing Trend in Prevalence of Gestational Diabetes Mellitus in a Large Population of Women between 1995 and 2005. Diabetes Care 2008, 31, 2288–2293. [Google Scholar] [CrossRef]
- Deitch, J.; Yates, C.J.; Hamblin, P.S.; Kevat, D.; Shahid, I.; Teale, G.; Lee, I. Prevalence of Gestational Diabetes Mellitus, Maternal Obesity and Associated Perinatal Outcomes over 10 Years in an Australian Tertiary Maternity Provider. Diabetes Res. Clin. Pract. 2023, 203, 110793. [Google Scholar] [CrossRef]
- Ferrara, A.; Kahn, H.S.; Quesenberry, C.P.; Riley, C.; Hedderson, M.M. An Increase in the Incidence of Gestational Diabetes Mellitus: Northern California, 1991–2000. Obstet. Gynecol. 2004, 103, 526–533. [Google Scholar] [CrossRef]
- Dabelea, D.; Snell-Bergeon, J.K.; Hartsfield, C.L.; Bischoff, K.J.; Hamman, R.F.; McDuffie, R.S.; Kaiser Permanente of Colorado GDM Screening Program. Increasing Prevalence of Gestational Diabetes Mellitus (GDM) over Time and by Birth Cohort: Kaiser Permanente of Colorado GDM Screening Program. Diabetes Care 2005, 28, 579–584. [Google Scholar] [CrossRef]
- Beischer, N.A.; Oats, J.N.; Henry, O.A.; Sheedy, M.T.; Walstab, J.E. Incidence and Severity of Gestational Diabetes Mellitus According to Country of Birth in Women Living in Australia. Diabetes 1991, 40 (Suppl. S2), 35–38. [Google Scholar] [CrossRef]
- Thorpe, L.E.; Berger, D.; Ellis, J.A.; Bettegowda, V.R.; Brown, G.; Matte, T.; Bassett, M.; Frieden, T.R. Trends and Racial/Ethnic Disparities in Gestational Diabetes Among Pregnant Women in New York City, 1990–2001. Am. J. Public Health 2005, 95, 1536–1539. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, C. Prevalence of Gestational Diabetes and Risk of Progression to Type 2 Diabetes: A Global Perspective. Curr. Diab. Rep. 2016, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, A. Increasing Prevalence of Gestational Diabetes Mellitus: A Public Health Perspective. Diabetes Care 2007, 30 (Suppl. S2), S141–S146. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. “Mendelian Randomization”: Can Genetic Epidemiology Contribute to Understanding Environmental Determinants of Disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef]
- Khankari, N.K.; Keaton, J.M.; Walker, V.M.; Lee, K.M.; Shuey, M.M.; Clarke, S.L.; Heberer, K.R.; Miller, D.R.; Reaven, P.D.; Lynch, J.A.; et al. Using Mendelian Randomisation to Identify Opportunities for Type 2 Diabetes Prevention by Repurposing Medications Used for Lipid Management. eBioMedicine 2022, 80, 104038. [Google Scholar] [CrossRef]
- Ji, Y.; Yiorkas, A.M.; Frau, F.; Mook-Kanamori, D.; Staiger, H.; Thomas, E.L.; Atabaki-Pasdar, N.; Campbell, A.; Tyrrell, J.; Jones, S.E.; et al. Genome-Wide and Abdominal MRI Data Provide Evidence That a Genetically Determined Favorable Adiposity Phenotype Is Characterized by Lower Ectopic Liver Fat and Lower Risk of Type 2 Diabetes, Heart Disease, and Hypertension. Diabetes 2019, 68, 207–219. [Google Scholar] [CrossRef]
- Swerdlow, D.I. Mendelian Randomization and Type 2 Diabetes. Cardiovasc. Drugs Ther. 2016, 30, 51–57. [Google Scholar] [CrossRef]
- Røder, M.E.; Porte, D.; Schwartz, R.S.; Kahn, S.E. Disproportionately Elevated Proinsulin Levels Reflect the Degree of Impaired B Cell Secretory Capacity in Patients with Noninsulin-Dependent Diabetes Mellitus. J. Clin. Endocrinol. Metab. 1998, 83, 604–608. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Hribal, M.L.; Oriente, F.; Accili, D. Mouse Models of Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E977–E981. [Google Scholar] [CrossRef][Green Version]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. Rev. 2019, 14, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 Diabetes as an Inflammatory Disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Duncan, B.B.; Schmidt, M.I.; Pankow, J.S.; Ballantyne, C.M.; Couper, D.; Vigo, A.; Hoogeveen, R.; Folsom, A.R.; Heiss, G. Low-Grade Systemic Inflammation and the Development of Type 2 Diabetes: The Atherosclerosis Risk in Communities Study. Diabetes 2003, 52, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-Reactive Protein, Interleukin 6, and Risk of Developing Type 2 Diabetes Mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.I.; Duncan, B.B.; Sharrett, A.R.; Lindberg, G.; Savage, P.J.; Offenbacher, S.; Azambuja, M.I.; Tracy, R.P.; Heiss, G. Markers of Inflammation and Prediction of Diabetes Mellitus in Adults (Atherosclerosis Risk in Communities Study): A Cohort Study. Lancet Lond. Engl. 1999, 353, 1649–1652. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and Inflammation: The Linking Mechanism and the Complications. Arch. Med. Sci. AMS 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Stępień, M.; Stępień, A.; Wlazeł, R.N.; Paradowski, M.; Banach, M.; Rysz, J. Obesity Indices and Inflammatory Markers in Obese Non-Diabetic Normo- and Hypertensive Patients: A Comparative Pilot Study. Lipids Health Dis. 2014, 13, 29. [Google Scholar] [CrossRef]
- Borges, M.C.; Clayton, G.L.; Freathy, R.M.; Felix, J.F.; Fernández-Sanlés, A.; Soares, A.G.; Kilpi, F.; Yang, Q.; McEachan, R.R.C.; Richmond, R.C.; et al. Integrating Multiple Lines of Evidence to Assess the Effects of Maternal BMI on Pregnancy and Perinatal Outcomes. BMC Med. 2024, 22, 32. [Google Scholar] [CrossRef]
- Pervjakova, N.; Moen, G.-H.; Borges, M.-C.; Ferreira, T.; Cook, J.P.; Allard, C.; Beaumont, R.N.; Canouil, M.; Hatem, G.; Heiskala, A.; et al. Multi-Ancestry Genome-Wide Association Study of Gestational Diabetes Mellitus Highlights Genetic Links with Type 2 Diabetes. Hum. Mol. Genet. 2022, 31, 3377–3391. [Google Scholar] [CrossRef]
- Shirazian, N.; Emdadi, R.; Mahboubi, M.; Motevallian, A.; Fazel-Sarjuei, Z.; Sedighpour, N.; Fadaki, S.-F.; Shahmoradi, N. Screening for Gestational Diabetes: Usefulness of Clinical Risk Factors. Arch. Gynecol. Obstet. 2009, 280, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Moses, R.; Griffiths, R.; Davis, W. Gestational Diabetes: Do All Women Need to Be Tested? Aust. N. Z. J. Obstet. Gynaecol. 1995, 35, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Lao, T.T.; Ho, L.-F.; Chan, B.C.P.; Leung, W.-C. Maternal Age and Prevalence of Gestational Diabetes Mellitus. Diabetes Care 2006, 29, 948–949. [Google Scholar] [CrossRef]
- Kim, C.; Berger, D.K.; Chamany, S. Recurrence of Gestational Diabetes Mellitus: A Systematic Review. Diabetes Care 2007, 30, 1314–1319. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, W.; Huang, W.; Zhang, L.; Liang, X.; Li, G. Differing Risk Factors for New Onset and Recurrent Gestational Diabetes Mellitus in Multipara Women: A Cohort Study. BMC Endocr. Disord. 2022, 22, 3. [Google Scholar] [CrossRef]
- Schwartz, N.; Nachum, Z.; Green, M.S. The Prevalence of Gestational Diabetes Mellitus Recurrence—Effect of Ethnicity and Parity: A Metaanalysis. Am. J. Obstet. Gynecol. 2015, 213, 310–317. [Google Scholar] [CrossRef]
- Larrabure-Torrealva, G.T.; Martinez, S.; Luque-Fernandez, M.A.; Sanchez, S.E.; Mascaro, P.A.; Ingar, H.; Castillo, W.; Zumaeta, R.; Grande, M.; Motta, V.; et al. Prevalence and Risk Factors of Gestational Diabetes Mellitus: Findings from a Universal Screening Feasibility Program in Lima, Peru. BMC Pregnancy Childbirth 2018, 18, 303. [Google Scholar] [CrossRef]
- Moosazadeh, M.; Asemi, Z.; Lankarani, K.B.; Tabrizi, R.; Maharlouei, N.; Naghibzadeh-Tahami, A.; Yousefzadeh, G.; Sadeghi, R.; Khatibi, S.R.; Afshari, M.; et al. Family History of Diabetes and the Risk of Gestational Diabetes Mellitus in Iran: A Systematic Review and Meta-Analysis. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, S99–S104. [Google Scholar] [CrossRef]
- Williams, M.A.; Qiu, C.; Dempsey, J.C.; Luthy, D.A. Familial Aggregation of Type 2 Diabetes and Chronic Hypertension in Women with Gestational Diabetes Mellitus. J. Reprod. Med. 2003, 48, 955–962. [Google Scholar]
- Hedderson, M.; Ehrlich, S.; Sridhar, S.; Darbinian, J.; Moore, S.; Ferrara, A. Racial/Ethnic Disparities in the Prevalence of Gestational Diabetes Mellitus by BMI. Diabetes Care 2012, 35, 1492–1498. [Google Scholar] [CrossRef]
- Kim, S.Y.; Saraiva, C.; Curtis, M.; Wilson, H.G.; Troyan, J.; Sharma, A.J. Fraction of Gestational Diabetes Mellitus Attributable to Overweight and Obesity by Race/Ethnicity, California, 2007–2009. Am. J. Public Health 2013, 103, e65–e72. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Esplin, E.D.; Oei, L.; Snyder, M.P. Personalized Sequencing and the Future of Medicine: Discovery, Diagnosis and Defeat of Disease. Pharmacogenomics 2014, 15, 1771–1790. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Lagou, V.; Welch, R.P.; Wheeler, E.; Montasser, M.E.; Luan, J.; Mägi, R.; Strawbridge, R.J.; Rehnberg, E.; Gustafsson, S.; et al. Large-Scale Association Analyses Identify New Loci Influencing Glycemic Traits and Provide Insight into the Underlying Biological Pathways. Nat. Genet. 2012, 44, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, E.; Leong, A.; Liu, C.-T.; Hivert, M.-F.; Strawbridge, R.J.; Podmore, C.; Li, M.; Yao, J.; Sim, X.; Hong, J.; et al. Impact of Common Genetic Determinants of Hemoglobin A1c on Type 2 Diabetes Risk and Diagnosis in Ancestrally Diverse Populations: A Transethnic Genome-Wide Meta-Analysis. PLOS Med. 2017, 14, e1002383. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Sidorenko, J.; Revez, J.A.; Xue, A.; Lu, X.; Pärna, K.; Snieder, H.; Visscher, P.M.; Wray, N.R.; Yengo, L. Estimation and Implications of the Genetic Architecture of Fasting and Non-Fasting Blood Glucose. Nat. Commun. 2023, 14, 451. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bakshi, A.; Zhu, Z.; Hemani, G.; Vinkhuyzen, A.A.E.; Lee, S.H.; Robinson, M.R.; Perry, J.R.B.; Nolte, I.M.; van Vliet-Ostaptchouk, J.V.; et al. Genetic Variance Estimation with Imputed Variants Finds Negligible Missing Heritability for Human Height and Body Mass Index. Nat. Genet. 2015, 47, 1114–1120. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.K.; Loh, P.-R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score Regression Distinguishes Confounding from Polygenicity in Genome-Wide Association Studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef]
- Moen, G.-H.; LeBlanc, M.; Sommer, C.; Prasad, R.B.; Lekva, T.; Normann, K.R.; Qvigstad, E.; Groop, L.; Birkeland, K.I.; Evans, D.M.; et al. Genetic Determinants of Glucose Levels in Pregnancy: Genetic Risk Scores Analysis and GWAS in the Norwegian STORK Cohort. Eur. J. Endocrinol. 2018, 179, 363–372. [Google Scholar] [CrossRef]
- Powe, C.E.; Nodzenski, M.; Talbot, O.; Allard, C.; Briggs, C.; Leya, M.V.; Perron, P.; Bouchard, L.; Florez, J.C.; Scholtens, D.M.; et al. Genetic Determinants of Glycemic Traits and the Risk of Gestational Diabetes Mellitus. Diabetes 2018, 67, 2703–2709. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007, 447, 661–678. Available online: https://pubmed.ncbi.nlm.nih.gov/17554300/ (accessed on 18 June 2024). [CrossRef] [PubMed]
- Zhen, J.; Gu, Y.; Wang, P.; Wang, W.; Bian, S.; Huang, S.; Liang, H.; Huang, M.; Yu, Y.; Chen, Q.; et al. Genome-Wide Association and Mendelian Randomisation Analysis among 30,699 Chinese Pregnant Women Identifies Novel Genetic and Molecular Risk Factors for Gestational Diabetes and Glycaemic Traits. Diabetologia 2024, 67, 703–713. [Google Scholar] [CrossRef]
- Wu, N.-N.; Zhao, D.; Ma, W.; Lang, J.-N.; Liu, S.-M.; Fu, Y.; Wang, X.; Wang, Z.-W.; Li, Q. A Genome-Wide Association Study of Gestational Diabetes Mellitus in Chinese Women. J. Matern.-Fetal Neonatal Med. 2021, 34, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Pei, L.; Lai, F.; Xiao, H.; Li, Z.; Zeng, R.; Chen, L.; Chen, W.; Liu, H.; Li, Y.; et al. Genome-Wide Analysis Study of Gestational Diabetes Mellitus and Related Pathogenic Factors in a Chinese Han Population. BMC Pregnancy Childbirth 2023, 23, 856. [Google Scholar] [CrossRef]
- Kwak, S.H.; Kim, S.H.; Cho, Y.M.; Go, M.J.; Cho, Y.S.; Choi, S.H.; Moon, M.K.; Jung, H.S.; Shin, H.D.; Kang, H.M.; et al. A Genome-Wide Association Study of Gestational Diabetes Mellitus in Korean Women. Diabetes 2012, 61, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.; Walters, R.K.; Pirinen, M.; Kurki, M.; Junna, N.; Goldstein, J.; Reeve, M.P.; Siirtola, H.; Lemmelä, S.; Turley, P.; et al. Distinct and Shared Genetic Architectures of Gestational Diabetes Mellitus and Type 2 Diabetes Mellitus. Nat. Genet. 2024, 56, 377–382. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef]
- Raimondo, A.; Rees, M.G.; Gloyn, A.L. Glucokinase Regulatory Protein: Complexity at the Crossroads of Triglyceride and Glucose Metabolism. Curr. Opin. Lipidol 2015, 26, 88–95. [Google Scholar] [CrossRef]
- Matschinsky, F.M.; Magnuson, M.A. Glucokinase and Glycemic Disease: From Basics to Novel Therapeutics; S.Karger AG: Basel, Switzerland, 2004. [Google Scholar] [CrossRef]
- Van Schaftingen, E.A. Protein from Rat Liver Confers to Glucokinase the Property of Being Antagonistically Regulated by Fructose 6-Phosphate and Fructose 1-Phosphate. Eur. J. Biochem. 1989, 179, 179–184. [Google Scholar] [CrossRef]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.V.; Orho-Melander, M.; Gloyn, A.L. The P446L Variant in GCKR Associated with Fasting Plasma Glucose and Triglyceride Levels Exerts Its Effect through Increased Glucokinase Activity in Liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef]
- Rees, M.G.; Wincovitch, S.; Schultz, J.; Waterstradt, R.; Beer, N.L.; Baltrusch, S.; Collins, F.S.; Gloyn, A.L. Cellular Characterisation of the GCKR P446L Variant Associated with Type 2 Diabetes Risk. Diabetologia 2012, 55, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Sparsø, T.; Andersen, G.; Nielsen, T.; Burgdorf, K.S.; Gjesing, A.P.; Nielsen, A.L.; Albrechtsen, A.; Rasmussen, S.S.; Jørgensen, T.; Borch-Johnsen, K.; et al. The GCKR Rs780094 Polymorphism Is Associated with Elevated Fasting Serum Triacylglycerol, Reduced Fasting and OGTT-Related Insulinaemia, and Reduced Risk of Type 2 Diabetes. Diabetologia 2008, 51, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research; Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.W.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; et al. Genome-Wide Association Analysis Identifies Loci for Type 2 Diabetes and Triglyceride Levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Onuma, H.; Tabara, Y.; Kawamoto, R.; Shimizu, I.; Kawamura, R.; Takata, Y.; Nishida, W.; Ohashi, J.; Miki, T.; Kohara, K.; et al. The GCKR Rs780094 Polymorphism Is Associated with Susceptibility of Type 2 Diabetes, Reduced Fasting Plasma Glucose Levels, Increased Triglycerides Levels and Lower HOMA-IR in Japanese Population. J. Hum. Genet. 2010, 55, 600–604. [Google Scholar] [CrossRef]
- Chen, G.; Shriner, D.; Zhang, J.; Zhou, J.; Adikaram, P.; Doumatey, A.P.; Bentley, A.R.; Adeyemo, A.; Rotimi, C.N. Additive Genetic Effect of GCKR, G6PC2, and SLC30A8 Variants on Fasting Glucose Levels and Risk of Type 2 Diabetes. PLoS ONE 2022, 17, e0269378. [Google Scholar] [CrossRef]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New Genetic Loci Implicated in Fasting Glucose Homeostasis and Their Impact on Type 2 Diabetes Risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef]
- Vaxillaire, M.; Cavalcanti-Proença, C.; Dechaume, A.; Tichet, J.; Marre, M.; Balkau, B.; Froguel, P.; for the DESIR Study Group. The Common P446L Polymorphism in GCKR Inversely Modulates Fasting Glucose and Triglyceride Levels and Reduces Type 2 Diabetes Risk in the DESIR Prospective General French Population. Diabetes 2008, 57, 2253–2257. [Google Scholar] [CrossRef]
- Lagou, V.; Jiang, L.; Ulrich, A.; Zudina, L.; González, K.S.G.; Balkhiyarova, Z.; Faggian, A.; Maina, J.G.; Chen, S.; Todorov, P.V.; et al. GWAS of Random Glucose in 476,326 Individuals Provide Insights into Diabetes Pathophysiology, Complications and Treatment Stratification. Nat. Genet. 2023, 55, 1448–1461. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Spracklen, C.N.; Marenne, G.; Varshney, A.; Corbin, L.J.; Luan, J.; Willems, S.M.; Wu, Y.; Zhang, X.; Horikoshi, M.; et al. The Trans-Ancestral Genomic Architecture of Glycemic Traits. Nat. Genet. 2021, 53, 840–860. [Google Scholar] [CrossRef]
- Sun, S.-C.; Lee, S.-E.; Xu, Y.-N.; Kim, N.-H. Perturbation of Spc25 Expression Affects Meiotic Spindle Organization, Chromosome Alignment and Spindle Assembly Checkpoint in Mouse Oocytes. Cell Cycle 2010, 9, 4552–4559. [Google Scholar] [CrossRef]
- Yon Jung, S.; Papp, J.C.; Sobel, E.M.; Pellegrini, M.; Yu, H. Genetic Variants of Glucose Metabolism and Exposure to Smoking in African American Breast Cancer. Endocr. Relat. Cancer 2023, 30, e220184. [Google Scholar] [CrossRef] [PubMed]
- Ellis, K.L.; Zhou, Y.; Beshansky, J.R.; Ainehsazan, E.; Yang, Y.; Selker, H.P.; Huggins, G.S.; Cupples, L.A.; Peter, I. Genetic Variation at Glucose and Insulin Trait Loci and Response to Glucose–Insulin–Potassium (GIK) Therapy: The IMMEDIATE Trial. Pharmacogenomics J. 2015, 15, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhu, Y.; Li, Z.; Bu, Q.; Sun, T.; Wang, H.; Sun, H.; Cao, X. Up-Regulation of SPC25 Promotes Breast Cancer. Aging 2019, 11, 5689–5704. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jia, J.; Huang, T. Shared Genetic Architecture and Casual Relationship between Leptin Levels and Type 2 Diabetes: Large-Scale Cross-Trait Meta-Analysis and Mendelian Randomization Analysis. BMJ Open Diabetes Res. Care 2020, 8, e001140. [Google Scholar] [CrossRef]
- Cui, F.; Hu, J.; Fan, Y.; Tan, J.; Tang, H. Knockdown of Spindle Pole Body Component 25 Homolog Inhibits Cell Proliferation and Cycle Progression in Prostate Cancer. Oncol. Lett. 2018, 15, 5712–5720. [Google Scholar] [CrossRef]
- Hutton, J.C.; O’Brien, R.M. Glucose-6-Phosphatase Catalytic Subunit Gene Family. J. Biol. Chem. 2009, 284, 29241–29245. [Google Scholar] [CrossRef]
- Arden, S.D.; Zahn, T.; Steegers, S.; Webb, S.; Bergman, B.; O’Brien, R.M.; Hutton, J.C. Molecular Cloning of a Pancreatic Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein. Diabetes 1999, 48, 531–542. [Google Scholar] [CrossRef]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef]
- Martin, C.C.; Bischof, L.J.; Bergman, B.; Hornbuckle, L.A.; Hilliker, C.; Frigeri, C.; Wahl, D.; Svitek, C.A.; Wong, R.; Goldman, J.K.; et al. Cloning and Characterization of the Human and Rat Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein (IGRP) Genes. J. Biol. Chem. 2001, 276, 25197–25207. [Google Scholar] [CrossRef]
- Mahajan, A.; Sim, X.; Ng, H.J.; Manning, A.; Rivas, M.A.; Highland, H.M.; Locke, A.E.; Grarup, N.; Im, H.K.; Cingolani, P.; et al. Identification and Functional Characterization of G6PC2 Coding Variants Influencing Glycemic Traits Define an Effector Transcript at the G6PC2-ABCB11 Locus. PLoS Genet. 2015, 11, e1004876. [Google Scholar] [CrossRef]
- Li, X.; Shu, Y.-H.; Xiang, A.H.; Trigo, E.; Kuusisto, J.; Hartiala, J.; Swift, A.J.; Kawakubo, M.; Stringham, H.M.; Bonnycastle, L.L.; et al. Additive Effects of Genetic Variation in GCK and G6PC2 on Insulin Secretion and Fasting Glucose. Diabetes 2009, 58, 2946–2953. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, Y.; Wang, J.; Wang, C.; Fan, J.; Zhao, J.; Yin, L.; Liu, X.; Zhang, D.; Li, L. Meta-Analyses of the Association of G6PC2 Allele Variants with Elevated Fasting Glucose and Type 2 Diabetes. PLoS ONE 2017, 12, e0181232. [Google Scholar] [CrossRef] [PubMed]
- Udhaya Kumar, S.; Kamaraj, B.; Varghese, R.P.; Preethi, V.A.; Bithia, R.; George Priya Doss, C. Mutations in G6PC2 Gene with Increased Risk for Development of Type 2 Diabetes: Understanding via Computational Approach. Adv. Protein Chem. Struct. Biol. 2022, 130, 351–373. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, J.; Querol, E.; Avilés, F.X. Metallocarboxypeptidases and Their Protein Inhibitors: Structure, Function and Biomedical Properties. Biochim. Biophys. Acta BBA—Protein Struct. Mol. Enzymol. 2000, 1477, 284–298. [Google Scholar] [CrossRef]
- Reznik, S.E.; Fricker, L.D. Carboxypeptidases from A to Z: Implications in Embryonic Development and Wnt Binding. Cell. Mol. Life Sci. CMLS 2001, 58, 1790–1804. [Google Scholar] [CrossRef]
- Lyons, P.J.; Fricker, L.D. Carboxypeptidase O Is a Glycosylphosphatidylinositol-Anchored Intestinal Peptidase with Acidic Amino Acid Specificity. J. Biol. Chem. 2011, 286, 39023–39032. [Google Scholar] [CrossRef] [PubMed]
- Beck, I.T. The Role of Pancreatic Enzymes in Digestion. Am. J. Clin. Nutr. 1973, 26, 311–325. [Google Scholar] [CrossRef]
- Burke, L.C.; Ezeribe, H.O.; Kwon, A.Y.; Dockery, D.; Lyons, P.J. Carboxypeptidase O Is a Lipid Droplet-Associated Enzyme Able to Cleave Both Acidic and Polar C-Terminal Amino Acids. PLoS ONE 2018, 13, e0206824. [Google Scholar] [CrossRef]
- Wei, S.; Segura, S.; Vendrell, J.; Aviles, F.X.; Lanoue, E.; Day, R.; Feng, Y.; Fricker, L.D. Identification and Characterization of Three Members of the Human Metallocarboxypeptidase Gene Family. J. Biol. Chem. 2002, 277, 14954–14964. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Taylor, A.J.; Fulcher, J.M.; Swensen, A.C.; Dai, X.-Q.; Komba, M.; Wrightson, K.L.C.; Fok, K.; Patterson, A.E.; Klein Geltink, R.I.; et al. Deletion of Carboxypeptidase E in β-Cells Disrupts Proinsulin Processing but Does Not Lead to Spontaneous Development of Diabetes in Mice. Diabetes 2023, 72, 1277–1288. [Google Scholar] [CrossRef]
- Alsters, S.I.M.; Goldstone, A.P.; Buxton, J.L.; Zekavati, A.; Sosinsky, A.; Yiorkas, A.M.; Holder, S.; Klaber, R.E.; Bridges, N.; van Haelst, M.M.; et al. Truncating Homozygous Mutation of Carboxypeptidase E (CPE) in a Morbidly Obese Female with Type 2 Diabetes Mellitus, Intellectual Disability and Hypogonadotrophic Hypogonadism. PLoS ONE 2015, 10, e0131417. [Google Scholar] [CrossRef]
- Naggert, J.K.; Fricker, L.D.; Varlamov, O.; Nishina, P.M.; Rouille, Y.; Steiner, D.F.; Carroll, R.J.; Paigen, B.J.; Leiter, E.H. Hyperproinsulinaemia in Obese Fat/Fat Mice Associated with a Carboxypeptidase E Mutation Which Reduces Enzyme Activity. Nat. Genet. 1995, 10, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Lockridge, A.; Alejandro, E.U. eIF4G1 and Carboxypeptidase E Axis Dysregulation in O-GlcNAc Transferase–Deficient Pancreatic β-Cells Contributes to Hyperproinsulinemia in Mice. J. Biol. Chem. 2019, 294, 13040–13050. [Google Scholar] [CrossRef] [PubMed]
- Defer, N.; Best-Belpomme, M.; Hanoune, J. Tissue Specificity and Physiological Relevance of Various Isoforms of Adenylyl Cyclase. Am. J. Physiol.-Ren. Physiol. 2000, 279, F400–F416. [Google Scholar] [CrossRef]
- Halls, M.L.; Cooper, D.M.F. Adenylyl Cyclase Signalling Complexes—Pharmacological Challenges and Opportunities. Pharmacol. Ther. 2017, 172, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Matschinsky, F.M. Ca2+, cAMP, and Phospholipid-Derived Messengers in Coupling Mechanisms of Insulin Secretion. Physiol. Rev. 1987, 67, 1185–1248. [Google Scholar] [CrossRef]
- Leech, C.A.; Castonguay, M.A.; Habener, J.F. Expression of Adenylyl Cyclase Subtypes in Pancreatic Beta-Cells. Biochem. Biophys. Res. Commun. 1999, 254, 703–706. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The Human Pancreatic Islet Transcriptome: Expression of Candidate Genes for Type 1 Diabetes and the Impact of pro-Inflammatory Cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 New Risk Loci for Type 2 Diabetes and Related Vascular Outcomes among 1.4 Million Participants in a Multi-Ancestry Meta-Analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Prasad, R.B.; Kristensen, K.; Katsarou, A.; Shaat, N. Association of Single Nucleotide Polymorphisms with Insulin Secretion, Insulin Sensitivity, and Diabetes in Women with a History of Gestational Diabetes Mellitus. BMC Med. Genomics 2021, 14, 274. [Google Scholar] [CrossRef]
- Saxena, R.; Hivert, M.-F.; Langenberg, C.; Tanaka, T.; Pankow, J.S.; Vollenweider, P.; Lyssenko, V.; Bouatia-Naji, N.; Dupuis, J.; Jackson, A.U.; et al. Genetic Variation in GIPR Influences the Glucose and Insulin Responses to an Oral Glucose Challenge. Nat. Genet. 2010, 42, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Huopio, H.; Cederberg, H.; Vangipurapu, J.; Hakkarainen, H.; Pääkkönen, M.; Kuulasmaa, T.; Heinonen, S.; Laakso, M. Association of Risk Variants for Type 2 Diabetes and Hyperglycemia with Gestational Diabetes. Eur. J. Endocrinol. 2013, 169, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Arora, G.P.; Almgren, P.; Brøns, C.; Thaman, R.G.; Vaag, A.A.; Groop, L.; Prasad, R.B. Association between Genetic Risk Variants and Glucose Intolerance during Pregnancy in North Indian Women. BMC Med. Genom. 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Scott, L.J.; Steinthorsdottir, V.; Morris, A.P.; Dina, C.; Welch, R.P.; Zeggini, E.; Huth, C.; Aulchenko, Y.S.; Thorleifsson, G.; et al. Twelve Type 2 Diabetes Susceptibility Loci Identified through Large-Scale Association Analysis. Nat. Genet. 2010, 42, 579–589. [Google Scholar] [CrossRef]
- Hodson, D.J.; Mitchell, R.K.; Marselli, L.; Pullen, T.J.; Gimeno Brias, S.; Semplici, F.; Everett, K.L.; Cooper, D.M.F.; Bugliani, M.; Marchetti, P.; et al. ADCY5 Couples Glucose to Insulin Secretion in Human Islets. Diabetes 2014, 63, 3009–3021. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Yuan, Z.; Zhang, C.; Ju, H.; Sun, Y.; Huang, N.; Chen, L.; Jin, L. Common Genetic Variants in ADCY5 and Gestational Glycemic Traits. PLoS ONE 2020, 15, e0230032. [Google Scholar] [CrossRef]
- Stijnen, P.; Ramos-Molina, B.; O’Rahilly, S.; Creemers, J.W.M. PCSK1 Mutations and Human Endocrinopathies: From Obesity to Gastrointestinal Disorders. Endocr. Rev. 2016, 37, 347–371. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Volders, K.; Stanhope, R.; Heuschkel, R.; White, A.; Lank, E.; Keogh, J.; O’Rahilly, S.; Creemers, J.W.M. Hyperphagia and Early-Onset Obesity Due to a Novel Homozygous Missense Mutation in Prohormone Convertase 1/3. J. Clin. Endocrinol. Metab. 2007, 92, 3369–3373. [Google Scholar] [CrossRef]
- Benjannet, S.; Rondeau, N.; Day, R.; Chrétien, M.; Seidah, N.G. PC1 and PC2 Are Proprotein Convertases Capable of Cleaving Proopiomelanocortin at Distinct Pairs of Basic Residues. Proc. Natl. Acad. Sci. USA 1991, 88, 3564–3568. [Google Scholar] [CrossRef]
- Thomas, L.; Leduc, R.; Thorne, B.A.; Smeekens, S.P.; Steiner, D.F.; Thomas, G. Kex2-like Endoproteases PC2 and PC3 Accurately Cleave a Model Prohormone in Mammalian Cells: Evidence for a Common Core of Neuroendocrine Processing Enzymes. Proc. Natl. Acad. Sci. USA 1991, 88, 5297–5301. [Google Scholar] [CrossRef]
- Dhanvantari, S.; Seidah, N.G.; Brubaker, P.L. Role of Prohormone Convertases in the Tissue-Specific Processing of Proglucagon. Mol. Endocrinol. Baltim. Md 1996, 10, 342–355. [Google Scholar] [CrossRef][Green Version]
- Rouillé, Y.; Kantengwa, S.; Irminger, J.C.; Halban, P.A. Role of the Prohormone Convertase PC3 in the Processing of Proglucagon to Glucagon-like Peptide 1. J. Biol. Chem. 1997, 272, 32810–32816. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Molina, B.; Martin, M.G.; Lindberg, I. PCSK1 Variants and Human Obesity. Prog. Mol. Biol. Transl. Sci. 2016, 140, 47–74. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Orci, L.; Carroll, R.; Norrbom, C.; Ravazzola, M.; Steiner, D.F. Severe Block in Processing of Proinsulin to Insulin Accompanied by Elevation of Des-64,65 Proinsulin Intermediates in Islets of Mice Lacking Prohormone Convertase 1/3. Proc. Natl. Acad. Sci. USA 2002, 99, 10299–10304. [Google Scholar] [CrossRef]
- Smeekens, S.P.; Montag, A.G.; Thomas, G.; Albiges-Rizo, C.; Carroll, R.; Benig, M.; Phillips, L.A.; Martin, S.; Ohagi, S.; Gardner, P. Proinsulin Processing by the Subtilisin-Related Proprotein Convertases Furin, PC2, and PC3. Proc. Natl. Acad. Sci. USA 1992, 89, 8822–8826. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, A.; Dey, A.; Norrbom, C.; Carroll, R.; Zhang, C.; Laurent, V.; Lindberg, I.; Ugleholdt, R.; Holst, J.J.; et al. Disruption of PC1/3 Expression in Mice Causes Dwarfism and Multiple Neuroendocrine Peptide Processing Defects. Proc. Natl. Acad. Sci. USA 2002, 99, 10293–10298. [Google Scholar] [CrossRef]
- Furuta, M.; Carroll, R.; Martin, S.; Swift, H.H.; Ravazzola, M.; Orci, L.; Steiner, D.F. Incomplete Processing of Proinsulin to Insulin Accompanied by Elevation of Des-31,32 Proinsulin Intermediates in Islets of Mice Lacking Active PC2. J. Biol. Chem. 1998, 273, 3431–3437. [Google Scholar] [CrossRef]
- Shen, W.-J.; Yao, T.; Kong, X.; Williams, K.W.; Liu, T. Melanocortin Neurons: Multiple Routes to Regulation of Metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863 Pt A, 2477–2485. [Google Scholar] [CrossRef]
- Yeo, G.S.H.; Chao, D.H.M.; Siegert, A.-M.; Koerperich, Z.M.; Ericson, M.D.; Simonds, S.E.; Larson, C.M.; Luquet, S.; Clarke, I.; Sharma, S.; et al. The Melanocortin Pathway and Energy Homeostasis: From Discovery to Obesity Therapy. Mol. Metab. 2021, 48, 101206. [Google Scholar] [CrossRef]
- Myers, M.G.; Olson, D.P. Central Nervous System Control of Metabolism. Nature 2012, 491, 357–363. [Google Scholar] [CrossRef]
- Yang, D.; Hou, X.; Yang, G.; Li, M.; Zhang, J.; Han, M.; Zhang, Y.; Liu, Y. Effects of the POMC System on Glucose Homeostasis and Potential Therapeutic Targets for Obesity and Diabetes. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Folon, L.; Baron, M.; Toussaint, B.; Vaillant, E.; Boissel, M.; Scherrer, V.; Loiselle, H.; Leloire, A.; Badreddine, A.; Balkau, B.; et al. Contribution of Heterozygous PCSK1 Variants to Obesity and Implications for Precision Medicine: A Case-Control Study. Lancet Diabetes Endocrinol. 2023, 11, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Löffler, D.; Behrendt, S.; Creemers, J.W.M.; Klammt, J.; Aust, G.; Stanik, J.; Kiess, W.; Kovacs, P.; Körner, A. Functional and Clinical Relevance of Novel and Known PCSK1 Variants for Childhood Obesity and Glucose Metabolism. Mol. Metab. 2017, 6, 295–305. [Google Scholar] [CrossRef]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-Wide Association Identifies Nine Common Variants Associated with Fasting Proinsulin Levels and Provides New Insights into the Pathophysiology of Type 2 Diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef]
- Nead, K.T.; Li, A.; Wehner, M.R.; Neupane, B.; Gustafsson, S.; Butterworth, A.; Engert, J.C.; Davis, A.D.; Hegele, R.A.; Miller, R.; et al. Contribution of Common Non-Synonymous Variants in PCSK1 to Body Mass Index Variation and Risk of Obesity: A Systematic Review and Meta-Analysis with Evidence from up to 331 175 Individuals. Hum. Mol. Genet. 2015, 24, 3582–3594. [Google Scholar] [CrossRef]
- Goyal, Y.; Verma, A.K.; Joshi, P.C.; Dev, K. Contemplating the Role of Genetic Variants of HHEX, CDKAL1, WFS1 and SLC30A8 Genes of TYPE-2 Diabetes in Asians Ethnic Groups. Gene Rep. 2019, 17, 100465. [Google Scholar] [CrossRef]
- Pierrel, F.; Douki, T.; Fontecave, M.; Atta, M. MiaB Protein Is a Bifunctional Radical-S-Adenosylmethionine Enzyme Involved in Thiolation and Methylation of tRNA. J. Biol. Chem. 2004, 279, 47555–47563. [Google Scholar] [CrossRef]
- Palmer, C.J.; Bruckner, R.J.; Paulo, J.A.; Kazak, L.; Long, J.Z.; Mina, A.I.; Deng, Z.; LeClair, K.B.; Hall, J.A.; Hong, S.; et al. Cdkal1, a Type 2 Diabetes Susceptibility Gene, Regulates Mitochondrial Function in Adipose Tissue. Mol. Metab. 2017, 6, 1212–1225. [Google Scholar] [CrossRef]
- El-Lebedy, D.; Ashmawy, I. Common Variants in TCF7L2 and CDKAL1 Genes and Risk of Type 2 Diabetes Mellitus in Egyptians. J. Genet. Eng. Biotechnol. 2016, 14, 247–251. [Google Scholar] [CrossRef]
- Steinthorsdottir, V.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Jonsdottir, T.; Walters, G.B.; Styrkarsdottir, U.; Gretarsdottir, S.; Emilsson, V.; Ghosh, S.; et al. A Variant in CDKAL1 Influences Insulin Response and Risk of Type 2 Diabetes. Nat. Genet. 2007, 39, 770–775. [Google Scholar] [CrossRef]
- Zeggini, E.; Weedon, M.N.; Lindgren, C.M.; Frayling, T.M.; Elliott, K.S.; Lango, H.; Timpson, N.J.; Perry, J.R.B.; Rayner, N.W.; Freathy, R.M.; et al. Replication of Genome-Wide Association Signals in UK Samples Reveals Risk Loci for Type 2 Diabetes. Science 2007, 316, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, L.; Tura, A.; Patel, S.K.; Ibrahim, I.M.; Ferrannini, E.; Zeggini, E.; Weedon, M.N.; Mari, A.; Hattersley, A.T.; McCarthy, M.I.; et al. Common Variants of the Novel Type 2 Diabetes Genes CDKAL1 and HHEX/IDE Are Associated with Decreased Pancreatic Beta-Cell Function. Diabetes 2007, 56, 3101–3104. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, H.A.; Madsen, A.L.; Viñuela, A.; Have, C.T.; Grarup, N.; Tura, A.; Mahajan, A.; Heggie, A.J.; Koivula, R.W.; De Masi, F.; et al. Genome-Wide Association Analysis of Pancreatic Beta-Cell Glucose Sensitivity. J. Clin. Endocrinol. Metab. 2020, 106, 80–90. [Google Scholar] [CrossRef]
- Ribas, V.; Drew, B.G.; Le, J.A.; Soleymani, T.; Daraei, P.; Sitz, D.; Mohammad, L.; Henstridge, D.C.; Febbraio, M.A.; Hewitt, S.C.; et al. Myeloid-Specific Estrogen Receptor Alpha Deficiency Impairs Metabolic Homeostasis and Accelerates Atherosclerotic Lesion Development. Proc. Natl. Acad. Sci. USA 2011, 108, 16457–16462. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef]
- Meoli, L.; Isensee, J.; Zazzu, V.; Nabzdyk, C.S.; Soewarto, D.; Witt, H.; Foryst-Ludwig, A.; Kintscher, U.; Noppinger, P.R. Sex- and Age-Dependent Effects of Gpr30 Genetic Deletion on the Metabolic and Cardiovascular Profiles of Diet-Induced Obese Mice. Gene 2014, 540, 210–216. [Google Scholar] [CrossRef]
- Barreto-Andrade, J.N.; de Fátima, L.A.; Campello, R.S.; Guedes, J.A.C.; de Freitas, H.S.; Machado, M.M.O.U.F. Estrogen Receptor 1 (ESR1) Enhances Slc2a4/GLUT4 Expression by a SP1 Cooperative Mechanism. Int. J. Med. Sci. 2018, 15, 1320–1328. [Google Scholar] [CrossRef]
- Gregorio, K.C.R.; Laurindo, C.P.; Machado, U.F. Estrogen and Glycemic Homeostasis: The Fundamental Role of Nuclear Estrogen Receptors ESR1/ESR2 in Glucose Transporter GLUT4 Regulation. Cells 2021, 10, 99. [Google Scholar] [CrossRef]
- Meyer, M.R.; Clegg, D.J.; Prossnitz, E.R.; Barton, M. Obesity, Insulin Resistance and Diabetes: Sex Differences and Role of Oestrogen Receptors. Acta Physiol. 2011, 203, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, I.; Vaxillaire, M.; Nilsson, M.; Lecoeur, C.; Gu, H.F.; Cavalcanti-Proença, C.; Efendic, S.; Ostenson, C.G.; Brismar, K.; Charpentier, G.; et al. Estrogen Receptor Alpha Gene Variants Associate with Type 2 Diabetes and Fasting Plasma Glucose. Pharmacogenet. Genom. 2008, 18, 967–975. [Google Scholar] [CrossRef]
- Fox, C.S.; Heard-Costa, N.; Cupples, L.A.; Dupuis, J.; Vasan, R.S.; Atwood, L.D. Genome-Wide Association to Body Mass Index and Waist Circumference: The Framingham Heart Study 100K Project. BMC Med. Genet. 2007, 8, S18. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.S.; Yang, Q.; Cupples, L.A.; Guo, C.-Y.; Atwood, L.D.; Murabito, J.M.; Levy, D.; Mendelsohn, M.E.; Housman, D.E.; Shearman, A.M. Sex-Specific Association between Estrogen Receptor-α Gene Variation and Measures of Adiposity: The Framingham Heart Study. J. Clin. Endocrinol. Metab. 2005, 90, 6257–6262. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, T.; Lu, W.; Mu, P.; Yang, Y.; Liang, W.; Li, C.; Lin, G. Estrogen Receptor Alpha Gene Polymorphism Associated with Type 2 Diabetes Mellitus and the Serum Lipid Concentration in Chinese Women in Guangzhou. Chin. Med. J. 2006, 119, 1794. [Google Scholar] [CrossRef] [PubMed]
- Ereqat, S.; Cauchi, S.; Eweidat, K.; Elqadi, M.; Nasereddin, A. Estrogen Receptor 1 Gene Polymorphisms (PvuII and XbaI) Are Associated with Type 2 Diabetes in Palestinian Women. PeerJ 2019, 7, e7164. [Google Scholar] [CrossRef]
- Lemaire, K.; Ravier, M.A.; Schraenen, A.; Creemers, J.W.M.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.A.; et al. Insulin Crystallization Depends on Zinc Transporter ZnT8 Expression, but Is Not Required for Normal Glucose Homeostasis in Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef]
- Chimienti, F.; Devergnas, S.; Pattou, F.; Schuit, F.; Garcia-Cuenca, R.; Vandewalle, B.; Kerr-Conte, J.; Van Lommel, L.; Grunwald, D.; Favier, A.; et al. In Vivo Expression and Functional Characterization of the Zinc Transporter ZnT8 in Glucose-Induced Insulin Secretion. J. Cell Sci. 2006, 119, 4199–4206. [Google Scholar] [CrossRef]
- Wijesekara, N.; Dai, F.F.; Hardy, A.B.; Giglou, P.R.; Bhattacharjee, A.; Koshkin, V.; Chimienti, F.; Gaisano, H.Y.; Rutter, G.A.; Wheeler, M.B. Beta Cell-Specific Znt8 Deletion in Mice Causes Marked Defects in Insulin Processing, Crystallisation and Secretion. Diabetologia 2010, 53, 1656–1668. [Google Scholar] [CrossRef]
- Tamaki, M.; Fujitani, Y.; Hara, A.; Uchida, T.; Tamura, Y.; Takeno, K.; Kawaguchi, M.; Watanabe, T.; Ogihara, T.; Fukunaka, A.; et al. The Diabetes-Susceptible Gene SLC30A8/ZnT8 Regulates Hepatic Insulin Clearance. J. Clin. Investig. 2013, 123, 4513–4524. [Google Scholar] [CrossRef]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.R.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-Function Mutations in SLC30A8 Protect against Type 2 Diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Bao, W.; Zhang, Y.; Rong, Y.; Wang, X.; Jin, Y.; Song, Y.; Yao, P.; Sun, C.; Hu, F.B.; et al. Interactions between Zinc Transporter-8 Gene (SLC30A8) and Plasma Zinc Concentrations for Impaired Glucose Regulation and Type 2 Diabetes. Diabetes 2014, 63, 1796–1803. [Google Scholar] [CrossRef]
- Xiang, J.; Li, X.-Y.; Xu, M.; Hong, J.; Huang, Y.; Tan, J.-R.; Lu, X.; Dai, M.; Yu, B.; Ning, G. Zinc Transporter-8 Gene (SLC30A8) Is Associated with Type 2 Diabetes in Chinese. J. Clin. Endocrinol. Metab. 2008, 93, 4107–4112. [Google Scholar] [CrossRef] [PubMed]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The Alternative Product from the Human CDKN2A Locus, P14(ARF), Participates in a Regulatory Feedback Loop with P53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of p16CDKN2 Expression and Its Implications for Cell Immortalization and Senescence. Mol. Cell. Biol. 1996, 16, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Flanders, T.Y.; Pollock, P.M.; Hayward, N.K. The CDKN2A (P16) Gene and Human Cancer. Mol. Med. 1997, 3, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Wouters, K.; Paumelle, R.; Staels, B. Functional Genomics of the CDKN2A/B Locus in Cardiovascular and Metabolic Disease: What Have We Learned from GWASs? Trends Endocrinol. Metab. 2015, 26, 176–184. [Google Scholar] [CrossRef]
- Kong, Y.; Sharma, R.B.; Ly, S.; Stamateris, R.E.; Jesdale, W.M.; Alonso, L.C. CDKN2A/B T2D Genome-Wide Association Study Risk SNPs Impact Locus Gene Expression and Proliferation in Human Islets. Diabetes 2018, 67, 872–884. [Google Scholar] [CrossRef]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A Genome-Wide Association Study of Type 2 Diabetes in Finns Detects Multiple Susceptibility Variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef]
- Irwin, D.M.; Tan, H. Molecular Evolution of the Vertebrate Hexokinase Gene Family: Identification of a Conserved Fifth Vertebrate Hexokinase Gene. Comp. Biochem. Physiol. Part D Genom. Proteom. 2008, 3, 96–107. [Google Scholar] [CrossRef]
- Khan, M.W.; Priyadarshini, M.; Cordoba-Chacon, J.; Becker, T.C.; Layden, B.T. Hepatic Hexokinase Domain Containing 1 (HKDC1) Improves Whole Body Glucose Tolerance and Insulin Sensitivity in Pregnant Mice. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 678–687. [Google Scholar] [CrossRef]
- Guo, C.; Ludvik, A.E.; Arlotto, M.E.; Hayes, M.G.; Armstrong, L.L.; Scholtens, D.M.; Brown, C.D.; Newgard, C.B.; Becker, T.C.; Layden, B.T.; et al. Coordinated Regulatory Variation Associated with Gestational Hyperglycaemia Regulates Expression of the Novel Hexokinase HKDC1. Nat. Commun. 2015, 6, 6069. [Google Scholar] [CrossRef]
- Hayes, M.G.; Urbanek, M.; Hivert, M.-F.; Armstrong, L.L.; Morrison, J.; Guo, C.; Lowe, L.P.; Scheftner, D.A.; Pluzhnikov, A.; Levine, D.M.; et al. Identification of HKDC1 and BACE2 as Genes Influencing Glycemic Traits during Pregnancy through Genome-Wide Association Studies. Diabetes 2013, 62, 3282–3291. [Google Scholar] [CrossRef]
- Kanthimathi, S.; Liju, S.; Laasya, D.; Anjana, R.M.; Mohan, V.; Radha, V. Hexokinase Domain Containing 1 (HKDC1) Gene Variants and Their Association with Gestational Diabetes Mellitus in a South Indian Population. Ann. Hum. Genet. 2016, 80, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-X.; Hu, S.-M.; You, Y.-P.; Yang, G.-L.; Wang, W. Replication of Previous Genome-Wide Association Studies of HKDC1, BACE2, SLC16A11 and TMEM163 SNPs in a Gestational Diabetes Mellitus Case-Control Sample from Han Chinese Population. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Zapater, J.L.; Lednovich, K.R.; Layden, B.T. The Role of Hexokinase Domain Containing Protein-1 in Glucose Regulation During Pregnancy. Curr. Diab. Rep. 2021, 21, 27. [Google Scholar] [CrossRef]
- Pusec, C.M.; Ilievski, V.; De Jesus, A.; Farooq, Z.; Zapater, J.L.; Sweis, N.; Ismail, H.; Khan, M.W.; Ardehali, H.; Cordoba-Chacon, J.; et al. Liver-Specific Overexpression of HKDC1 Increases Hepatocyte Size and Proliferative Capacity. Sci. Rep. 2023, 13, 8034. [Google Scholar] [CrossRef]
- Zhou, Y.; Park, S.-Y.; Su, J.; Bailey, K.; Ottosson-Laakso, E.; Shcherbina, L.; Oskolkov, N.; Zhang, E.; Thevenin, T.; Fadista, J.; et al. TCF7L2 Is a Master Regulator of Insulin Production and Processing. Hum. Mol. Genet. 2014, 23, 6419–6431. [Google Scholar] [CrossRef] [PubMed]
- Osmark, P.; Hansson, O.; Jonsson, A.; Rönn, T.; Groop, L.; Renström, E. Unique Splicing Pattern of the TCF7L2 Gene in Human Pancreatic Islets. Diabetologia 2009, 52, 850–854. [Google Scholar] [CrossRef]
- Wu, H.-H.; Li, Y.-L.; Liu, N.-J.; Yang, Z.; Tao, X.-M.; Du, Y.-P.; Wang, X.-C.; Lu, B.; Zhang, Z.-Y.; Hu, R.-M.; et al. TCF7L2 Regulates Pancreatic β-Cell Function through PI3K/AKT Signal Pathway. Diabetol. Metab. Syndr. 2019, 11, 55. [Google Scholar] [CrossRef]
- Grant, S.F.A.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of Transcription Factor 7-like 2 (TCF7L2) Gene Confers Risk of Type 2 Diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef]
- Dou, H.; Ma, E.; Yin, L.; Jin, Y.; Wang, H. The Association between Gene Polymorphism of TCF7L2 and Type 2 Diabetes in Chinese Han Population: A Meta-Analysis. PLoS ONE 2013, 8, e59495. [Google Scholar] [CrossRef]
- Qiao, H.; Zhang, X.; Zhao, X.; Zhao, Y.; Xu, L.; Sun, H.; Fu, S. Genetic Variants of TCF7L2 Are Associated with Type 2 Diabetes in a Northeastern Chinese Population. Gene 2012, 495, 115–119. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhao, J.; You, H.; Pang, C.; Yin, L.; Guo, T.; Feng, T.; Wang, C.; Gao, K.; Luo, X.; et al. Association of the Rs11196218 Polymorphism in TCF7L2 with Type 2 Diabetes Mellitus in Asian Population. Meta Gene 2014, 2, 332–341. [Google Scholar] [CrossRef]
- Gloyn, A.L.; Braun, M.; Rorsman, P. Type 2 Diabetes Susceptibility Gene TCF7L2 and Its Role in β-Cell Function. Diabetes 2009, 58, 800–802. [Google Scholar] [CrossRef]
- Keaton, J.M.; Gao, C.; Guan, M.; Hellwege, J.N.; Palmer, N.D.; Pankow, J.S.; Fornage, M.; Wilson, J.G.; Correa, A.; Rasmussen-Torvik, L.J.; et al. Genome-Wide Interaction with the Insulin Secretion Locus MTNR1B Reveals CMIP as a Novel Type 2 Diabetes Susceptibility Gene in African Americans. Genet. Epidemiol. 2018, 42, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.C.; da Silva, M.E.R.; Fukui, R.T.; Arruda-Marques, M.d.C.; dos Santos, R.F. TCF7L2 Correlation in Both Insulin Secretion and Postprandial Insulin Sensitivity. Diabetol. Metab. Syndr. 2018, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zhu, Y.; Lü, B.; Xu, F.; Li, X.; Lai, M. TCF7L2 Gene Polymorphisms and Type 2 Diabetes Risk: A Comprehensive and Updated Meta-Analysis Involving 121 174 Subjects. Mutagenesis 2013, 28, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Le Bacquer, O.; Kerr-Conte, J.; Gargani, S.; Delalleau, N.; Huyvaert, M.; Gmyr, V.; Froguel, P.; Neve, B.; Pattou, F. TCF7L2 Rs7903146 Impairs Islet Function and Morphology in Non-Diabetic Individuals. Diabetologia 2012, 55, 2677–2681. [Google Scholar] [CrossRef]
- Tuomi, T.; Nagorny, C.L.F.; Singh, P.; Bennet, H.; Yu, Q.; Alenkvist, I.; Isomaa, B.; Östman, B.; Söderström, J.; Pesonen, A.-K.; et al. Increased Melatonin Signaling Is a Risk Factor for Type 2 Diabetes. Cell Metab. 2016, 23, 1067–1077. [Google Scholar] [CrossRef]
- McMullan, C.J.; Schernhammer, E.S.; Rimm, E.B.; Hu, F.B.; Forman, J.P. Melatonin Secretion and the Incidence of Type 2 Diabetes. JAMA 2013, 309, 1388–1396. [Google Scholar] [CrossRef]
- Stumpf, I.; Mühlbauer, E.; Peschke, E. Involvement of the cGMP Pathway in Mediating the Insulin-Inhibitory Effect of Melatonin in Pancreatic β-Cells. J. Pineal Res. 2008, 45, 318–327. [Google Scholar] [CrossRef]
- Bass, J.; Takahashi, J.S. Circadian Integration of Metabolism and Energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef]
- Scheer, F.A.J.L.; Hilton, M.F.; Mantzoros, C.S.; Shea, S.A. Adverse Metabolic and Cardiovascular Consequences of Circadian Misalignment. Proc. Natl. Acad. Sci. USA 2009, 106, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-F.; Zhang, J.; Zhang, M.-Z.; Ni, J.; Wang, T.; Zhao, Y.-Q.; Khan, N.U. Evaluation of the Effect of MTNR1B Rs10830963 Gene Variant on the Therapeutic Efficacy of Nateglinide in Treating Type 2 Diabetes among Chinese Han Patients. BMC Med. Genomics 2021, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Nagorny, C.L.F.; Erdos, M.R.; Wierup, N.; Jonsson, A.; Spégel, P.; Bugliani, M.; Saxena, R.; Fex, M.; Pulizzi, N.; et al. Common Variant in MTNR1B Associated with Increased Risk of Type 2 Diabetes and Impaired Early Insulin Secretion. Nat. Genet. 2009, 41, 82–88. [Google Scholar] [CrossRef]
- Langenberg, C.; Pascoe, L.; Mari, A.; Tura, A.; Laakso, M.; Frayling, T.M.; Barroso, I.; Loos, R.J.F.; Wareham, N.J.; Walker, M.; et al. Common Genetic Variation in the Melatonin Receptor 1B Gene (MTNR1B) Is Associated with Decreased Early-Phase Insulin Response. Diabetologia 2009, 52, 1537–1542. [Google Scholar] [CrossRef]
- Liu, C.; Wu, Y.; Li, H.; Qi, Q.; Langenberg, C.; Loos, R.J.; Lin, X. MTNR1B Rs10830963 Is Associated with Fasting Plasma Glucose, HbA1Cand Impaired Beta-Cell Function in Chinese Hans from Shanghai. BMC Med. Genet. 2010, 11, 59. [Google Scholar] [CrossRef]
- Prokopenko, I.; Langenberg, C.; Florez, J.C.; Saxena, R.; Soranzo, N.; Thorleifsson, G.; Loos, R.J.F.; Manning, A.K.; Jackson, A.U.; Aulchenko, Y.; et al. Variants in MTNR1B Influence Fasting Glucose Levels. Nat. Genet. 2009, 41, 77–81. [Google Scholar] [CrossRef]
- Vejrazkova, D.; Vankova, M.; Vcelak, J.; Krejci, H.; Anderlova, K.; Tura, A.; Pacini, G.; Sumova, A.; Sladek, M.; Bendlova, B. The Rs10830963 Polymorphism of the MTNR1B Gene: Association With Abnormal Glucose, Insulin and C-Peptide Kinetics. Front. Endocrinol. 2022, 13, 868364. [Google Scholar] [CrossRef]
- Bonnefond, A.; Clément, N.; Fawcett, K.; Yengo, L.; Vaillant, E.; Guillaume, J.-L.; Dechaume, A.; Payne, F.; Roussel, R.; Czernichow, S.; et al. Rare MTNR1B Variants Impairing Melatonin Receptor 1B Function Contribute to Type 2 Diabetes. Nat. Genet. 2012, 44, 297–301. [Google Scholar] [CrossRef]
- Been, L.F.; Hatfield, J.L.; Shankar, A.; Aston, C.E.; Ralhan, S.; Wander, G.S.; Mehra, N.K.; Singh, J.R.; Mulvihill, J.J.; Sanghera, D.K. A Low Frequency Variant within the GWAS Locus of MTNR1B Affects Fasting Glucose Concentrations: Genetic Risk Is Modulated by Obesity. Nutr. Metab. Cardiovasc. Dis. NMCD 2012, 22, 944–951. [Google Scholar] [CrossRef]
- Wang, T.; Wang, X.; Lai, R.; Ling, H.; Zhang, F.; Lu, Q.; Lv, D.; Yin, X. MTNR1B Gene Polymorphisms Are Associated With the Therapeutic Responses to Repaglinide in Chinese Patients With Type 2 Diabetes Mellitus. Front. Pharmacol. 2019, 10, 1318. [Google Scholar] [CrossRef] [PubMed]
- Müssig, K.; Staiger, H.; Machicao, F.; Häring, H.-U.; Fritsche, A. Genetic Variants in MTNR1B Affecting Insulin Secretion. Ann. Med. 2010, 42, 387–393. [Google Scholar] [CrossRef]
- Kumar, S.; Matsuzaki, T.; Yoshida, Y.; Noda, M. Molecular Cloning and Biological Activity of a Novel Developmentally Regulated Gene Encoding a Protein with Beta-Transducin-like Structure. J. Biol. Chem. 1994, 269, 11318–11326. [Google Scholar] [CrossRef]
- Lüders, J.; Patel, U.K.; Stearns, T. GCP-WD Is a γ-Tubulin Targeting Factor Required for Centrosomal and Chromatin-Mediated Microtubule Nucleation. Nat. Cell Biol. 2006, 8, 137–147. [Google Scholar] [CrossRef]
- Haren, L.; Remy, M.-H.; Bazin, I.; Callebaut, I.; Wright, M.; Merdes, A. NEDD1-Dependent Recruitment of the γ-Tubulin Ring Complex to the Centrosome Is Necessary for Centriole Duplication and Spindle Assembly. J. Cell Biol. 2006, 172, 505–515. [Google Scholar] [CrossRef]
- Manning, J.A.; Shalini, S.; Risk, J.M.; Day, C.L.; Kumar, S. A Direct Interaction with NEDD1 Regulates γ-Tubulin Recruitment to the Centrosome. PLoS ONE 2010, 5, e9618. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Mammalian Cyclin-Dependent Kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To Cycle or Not to Cycle: A Critical Decision in Cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- Muñiz, L.C.; Yehia, G.; Mémin, E.; Ratnakar, P.V.A.L.; Molina, C.A. Transcriptional Regulation of Cyclin D2 by the PKA Pathway and Inducible cAMP Early Repressor in Granulosa Cells. Biol. Reprod. 2006, 75, 279–288. [Google Scholar] [CrossRef]
- Robker, R.L.; Richards, J.S. Hormone-Induced Proliferation and Differentiation of Granulosa Cells: A Coordinated Balance of the Cell Cycle Regulators Cyclin D2 and p27Kip1. Mol. Endocrinol. 1998, 12, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Quelle, D.E.; Ashmun, R.A.; Shurtleff, S.A.; Kato, J.Y.; Bar-Sagi, D.; Roussel, M.F.; Sherr, C.J. Overexpression of Mouse D-Type Cyclins Accelerates G1 Phase in Rodent Fibroblasts. Genes Dev. 1993, 7, 1559–1571. [Google Scholar] [CrossRef]
- Han, Y.; Xia, G.; Tsang, B.K. Regulation of Cyclin D2 Expression and Degradation by Follicle-Stimulating Hormone During Rat Granulosa Cell Proliferation In Vitro1. Biol. Reprod. 2013, 88, 57. [Google Scholar] [CrossRef] [PubMed]
- Salpeter, S.J.; Klochendler, A.; Weinberg-Corem, N.; Porat, S.; Granot, Z.; Shapiro, A.M.J.; Magnuson, M.A.; Eden, A.; Grimsby, J.; Glaser, B.; et al. Glucose Regulates Cyclin D2 Expression in Quiescent and Replicating Pancreatic β-Cells through Glycolysis and Calcium Channels. Endocrinology 2011, 152, 2589–2598. [Google Scholar] [CrossRef]
- McDermott, J.H.; Hickson, N.; Banerjee, I.; Murray, P.g.; Ram, D.; Metcalfe, K.; Clayton-Smith, J.; Douzgou, S. Hypoglycaemia Represents a Clinically Significant Manifestation of PIK3CA- and CCND2-Associated Segmental Overgrowth. Clin. Genet. 2018, 93, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Georgia, S.; Hinault, C.; Kawamori, D.; Hu, J.; Meyer, J.; Kanji, M.; Bhushan, A.; Kulkarni, R.N. Cyclin D2 Is Essential for the Compensatory β-Cell Hyperplastic Response to Insulin Resistance in Rodents. Diabetes 2010, 59, 987–996. [Google Scholar] [CrossRef]
- Yaghootkar, H.; Stancáková, A.; Freathy, R.M.; Vangipurapu, J.; Weedon, M.N.; Xie, W.; Wood, A.R.; Ferrannini, E.; Mari, A.; Ring, S.M.; et al. Association Analysis of 29,956 Individuals Confirms That a Low-Frequency Variant at CCND2 Halves the Risk of Type 2 Diabetes by Enhancing Insulin Secretion. Diabetes 2015, 64, 2279–2285. [Google Scholar] [CrossRef]
- Grimbert, P.; Valanciute, A.; Audard, V.; Pawlak, A.; Le Gouvelo, S.; Lang, P.; Niaudet, P.; Bensman, A.; Guellaën, G.; Sahali, D. Truncation of C-Mip (Tc-Mip), a New Proximal Signaling Protein, Induces c-Maf Th2 Transcription Factor and Cytoskeleton Reorganization. J. Exp. Med. 2003, 198, 797–807. [Google Scholar] [CrossRef]
- Kamal, M.; Valanciute, A.; Dahan, K.; Ory, V.; Pawlak, A.; Lang, P.; Guellaen, G.; Sahali, D. C-Mip Interacts Physically with RelA and Inhibits Nuclear Factor Kappa B Activity. Mol. Immunol. 2009, 46, 991–998. [Google Scholar] [CrossRef]
- Ollero, M.; Sahali, D. The Enigmatic Emerging Role of the C-Maf Inducing Protein in Cancer. Diagnostics 2021, 11, 666. [Google Scholar] [CrossRef]
- Scott, R.A.; Scott, L.J.; Mägi, R.; Marullo, L.; Gaulton, K.J.; Kaakinen, M.; Pervjakova, N.; Pers, T.H.; Johnson, A.D.; Eicher, J.D.; et al. An Expanded Genome-Wide Association Study of Type 2 Diabetes in Europeans. Diabetes 2017, 66, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, I.; Rydén, M.; Brodin, D.; Grallert, H.; Strawbridge, R.J.; Arner, P. Numerous Genes in Loci Associated With Body Fat Distribution Are Linked to Adipose Function. Diabetes 2016, 65, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Imamura, M.; Tanaka, Y.; Iwata, M.; Hirose, H.; Kaku, K.; Maegawa, H.; Watada, H.; Tobe, K.; Kashiwagi, A.; et al. Replication Study for the Association of 9 East Asian GWAS-Derived Loci with Susceptibility to Type 2 Diabetes in a Japanese Population. PLoS ONE 2013, 8, e76317. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, T.; Wu, Y.; Juan, J.; Qin, X.; Tang, X.; Wu, T.; Hu, Y. Opposite Genetic Effects of CMIP Polymorphisms on the Risk of Type 2 Diabetes and Obesity: A Family-Based Study in China. Int. J. Mol. Sci. 2018, 19, 1011. [Google Scholar] [CrossRef]
- Zhao, W.; Rasheed, A.; Tikkanen, E.; Lee, J.-J.; Butterworth, A.S.; Howson, J.M.M.; Assimes, T.L.; Chowdhury, R.; Orho-Melander, M.; Damrauer, S.; et al. Identification of New Susceptibility Loci for Type 2 Diabetes and Shared Etiological Pathways with Coronary Heart Disease. Nat. Genet. 2017, 49, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Mägi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New Genetic Loci Link Adipose and Insulin Biology to Body Fat Distribution. Nature 2015, 518, 187–196. [Google Scholar] [CrossRef]
- Wen, W.; Kato, N.; Hwang, J.-Y.; Guo, X.; Tabara, Y.; Li, H.; Dorajoo, R.; Yang, X.; Tsai, F.-J.; Li, S.; et al. Genome-Wide Association Studies in East Asians Identify New Loci for Waist-Hip Ratio and Waist Circumference. Sci. Rep. 2016, 6, 17958. [Google Scholar] [CrossRef]
- Yan, Y.-X.; Li, J.-J.-H.; Xiao, H.-B.; Wang, S.; He, Y.; Wu, L.-J. Association Analysis of Copy Number Variations in Type 2 Diabetes-Related Susceptible Genes in a Chinese Population. Acta Diabetol. 2018, 55, 909–916. [Google Scholar] [CrossRef]
- Cho, Y.S.; Chen, C.-H.; Hu, C.; Long, J.; Hee Ong, R.T.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-Analysis of Genome-Wide Association Studies Identifies Eight New Loci for Type 2 Diabetes in East Asians. Nat. Genet. 2012, 44, 67–72. [Google Scholar] [CrossRef]
- Hattori, K.; Naguro, I.; Runchel, C.; Ichijo, H. The Roles of ASK Family Proteins in Stress Responses and Diseases. Cell Commun. Signal. CCS 2009, 7, 9. [Google Scholar] [CrossRef]
- Kaji, T.; Yoshida, S.; Kawai, K.; Fuchigami, Y.; Watanabe, W.; Kubodera, H.; Kishimoto, T. ASK3, a Novel Member of the Apoptosis Signal-Regulating Kinase Family, Is Essential for Stress-Induced Cell Death in HeLa Cells. Biochem. Biophys. Res. Commun. 2010, 395, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-Activated Protein Kinase: Conservation of a Three-Kinase Module from Yeast to Human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Noguchi, T.; Naguro, I.; Ichijo, H. Apoptosis Signal-Regulating Kinase 1 in Stress and Immune Response. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 199–225. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, M.; Naguro, I.; Ichijo, H. In Vivo Gene Manipulation Reveals the Impact of Stress-Responsive MAPK Pathways on Tumor Progression. Cancer Sci. 2015, 106, 785–796. [Google Scholar] [CrossRef]
- Nag, A.; Dhindsa, R.S.; Mitchell, J.; Vasavda, C.; Harper, A.R.; Vitsios, D.; Ahnmark, A.; Bilican, B.; Madeyski-Bengtson, K.; Zarrouki, B.; et al. Human Genetics Uncovers MAP3K15 as an Obesity-Independent Therapeutic Target for Diabetes. Sci. Adv. 2022, 8, eadd5430. [Google Scholar] [CrossRef]
- Wang, Q.; Dhindsa, R.S.; Carss, K.; Harper, A.R.; Nag, A.; Tachmazidou, I.; Vitsios, D.; Deevi, S.V.V.; Mackay, A.; Muthas, D.; et al. Rare Variant Contribution to Human Disease in 281,104 UK Biobank Exomes. Nature 2021, 597, 527–532. [Google Scholar] [CrossRef]
- Backman, J.D.; Li, A.H.; Marcketta, A.; Sun, D.; Mbatchou, J.; Kessler, M.D.; Benner, C.; Liu, D.; Locke, A.E.; Balasubramanian, S.; et al. Exome Sequencing and Analysis of 454,787 UK Biobank Participants. Nature 2021, 599, 628–634. [Google Scholar] [CrossRef]
- Foley, C.N.; Staley, J.R.; Breen, P.G.; Sun, B.B.; Kirk, P.D.W.; Burgess, S.; Howson, J.M.M. A Fast and Efficient Colocalization Algorithm for Identifying Shared Genetic Risk Factors across Multiple Traits. Nat. Commun. 2021, 12, 764. [Google Scholar] [CrossRef]
- Farrar, D.; Fairley, L.; Santorelli, G.; Tuffnell, D.; Sheldon, T.A.; Wright, J.; van Overveld, L.; Lawlor, D.A. Association between Hyperglycaemia and Adverse Perinatal Outcomes in South Asian and White British Women: Analysis of Data from the Born in Bradford Cohort. Lancet Diabetes Endocrinol. 2015, 3, 795–804. [Google Scholar] [CrossRef]
- Suzuki, K.; Hatzikotoulas, K.; Southam, L.; Taylor, H.J.; Yin, X.; Lorenz, K.M.; Mandla, R.; Huerta-Chagoya, A.; Rayner, N.W.; Bocher, O.; et al. Multi-Ancestry Genome-Wide Study in >2.5 Million Individuals Reveals Heterogeneity in Mechanistic Pathways of Type 2 Diabetes and Complications. medRxiv 2023. [Google Scholar] [CrossRef]
- Mahajan, A.; Spracklen, C.N.; Zhang, W.; Ng, M.C.Y.; Petty, L.E.; Kitajima, H.; Yu, G.Z.; Rüeger, S.; Speidel, L.; Kim, Y.J.; et al. Multi-Ancestry Genetic Study of Type 2 Diabetes Highlights the Power of Diverse Populations for Discovery and Translation. Nat. Genet. 2022, 54, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Bryson, C.L.; Ioannou, G.N.; Rulyak, S.J.; Critchlow, C. Association between Gestational Diabetes and Pregnancy-Induced Hypertension. Am. J. Epidemiol. 2003, 158, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Guillén, M.A.; Herranz, L.; Barquiel, B.; Hillman, N.; Burgos, M.A.; Pallardo, L.F. Influence of Gestational Diabetes Mellitus on Neonatal Weight Outcome in Twin Pregnancies. Diabet. Med. 2014, 31, 1651–1656. [Google Scholar] [CrossRef]
- Joffe, G.M.; Esterlitz, J.R.; Levine, R.J.; Clemens, J.D.; Ewell, M.G.; Sibai, B.M.; Catalano, P.M. The Relationship between Abnormal Glucose Tolerance and Hypertensive Disorders of Pregnancy in Healthy Nulliparous Women. Calcium for Preeclampsia Prevention (CPEP) Study Group. Am. J. Obstet. Gynecol. 1998, 179, 1032–1037. [Google Scholar] [CrossRef]
- Gorgal, R.; Gonçalves, E.; Barros, M.; Namora, G.; Magalhães, A.; Rodrigues, T.; Montenegro, N. Gestational Diabetes Mellitus: A Risk Factor for Non-Elective Cesarean Section. J. Obstet. Gynaecol. Res. 2012, 38, 154–159. [Google Scholar] [CrossRef]
- Phaloprakarn, C.; Tangjitgamol, S. Risk Score for Predicting Primary Cesarean Delivery in Women with Gestational Diabetes Mellitus. BMC Pregnancy Childbirth 2020, 20, 607. [Google Scholar] [CrossRef]
- Persson, M.; Norman, M.; Hanson, U. Obstetric and Perinatal Outcomes in Type 1 Diabetic Pregnancies: A Large, Population-Based Study. Diabetes Care 2009, 32, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Farrar, D.; Simmonds, M.; Bryant, M.; Sheldon, T.A.; Tuffnell, D.; Golder, S.; Dunne, F.; Lawlor, D.A. Hyperglycaemia and Risk of Adverse Perinatal Outcomes: Systematic Review and Meta-Analysis. Obstet. Anesth. Dig. 2017, 37, 64. [Google Scholar] [CrossRef]
- Boriboonhirunsarn, D.; Tanpong, S. Rate of Spontaneous Preterm Delivery Between Pregnant Women With and Without Gestational Diabetes. Cureus 2023, 15, e34565. [Google Scholar] [CrossRef]
- González González, N.L.; Goya, M.; Bellart, J.; Lopez, J.; Sancho, M.A.; Mozas, J.; Medina, V.; Padrón, E.; Megia, A.; Pintado, P.; et al. Obstetric and Perinatal Outcome in Women with Twin Pregnancy and Gestational Diabetes. J. Matern. Fetal Neonatal Med. 2012, 25, 1084–1089. [Google Scholar] [CrossRef]
- Balsells, M.; García-Patterson, A.; Gich, I.; Corcoy, R. Maternal and Fetal Outcome in Women with Type 2 Versus Type 1 Diabetes Mellitus: A Systematic Review and Metaanalysis. J. Clin. Endocrinol. Metab. 2009, 94, 4284–4291. [Google Scholar] [CrossRef] [PubMed]
- Murphy, H.R.; Steel, S.A.; Roland, J.M.; Morris, D.; Ball, V.; Campbell, P.J.; Temple, R.C.; East Anglia Study Group for Improving Pregnancy Outcomes in Women with Diabetes (EASIPOD). Obstetric and Perinatal Outcomes in Pregnancies Complicated by Type 1 and Type 2 Diabetes: Influences of Glycaemic Control, Obesity and Social Disadvantage. Diabet. Med. 2011, 28, 1060–1067. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, B.; Sun, Y.; Du, Y.; Santillan, M.K.; Santillan, D.A.; Snetselaar, L.G.; Bao, W. Association of Maternal Prepregnancy Diabetes and Gestational Diabetes Mellitus With Congenital Anomalies of the Newborn. Diabetes Care 2020, 43, 2983–2990. [Google Scholar] [CrossRef] [PubMed]
- Greco, E.; Calanducci, M.; Nicolaides, K.H.; Barry, E.V.H.; Huda, M.S.B.; Iliodromiti, S. Gestational Diabetes Mellitus and Adverse Maternal and Perinatal Outcomes in Twin and Singleton Pregnancies: A Systematic Review and Meta-Analysis. Am. J. Obstet. Gynecol. 2024, 230, 213–225. [Google Scholar] [CrossRef]
- Ye, W.; Luo, C.; Huang, J.; Li, C.; Liu, Z.; Liu, F. Gestational Diabetes Mellitus and Adverse Pregnancy Outcomes: Systematic Review and Meta-Analysis. BMJ 2022, 377, e067946. [Google Scholar] [CrossRef] [PubMed]
- HAPO Study Cooperative Research Group; Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; et al. Hyperglycemia and Adverse Pregnancy Outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [CrossRef]
- McBride, N.; Fernández-Sanlés, A.; Arab, M.A.; Bond, T.A.; Zheng, J.; Magnus, M.C.; Corfield, E.C.; Clayton, G.L.; Hwang, L.-D.; Beaumont, R.N.; et al. Effects of the Maternal and Fetal Proteome on Birth Weight: A Mendelian Randomization Analysis. medRxiv 2023. [Google Scholar] [CrossRef]
- Song, C.; Lyu, Y.; Li, C.; Liu, P.; Li, J.; Ma, R.C.; Yang, X. Long-Term Risk of Diabetes in Women at Varying Durations after Gestational Diabetes: A Systematic Review and Meta-Analysis with More than 2 Million Women. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2018, 19, 421–429. [Google Scholar] [CrossRef]
- Vounzoulaki, E.; Khunti, K.; Abner, S.C.; Tan, B.K.; Davies, M.J.; Gillies, C.L. Progression to Type 2 Diabetes in Women with a Known History of Gestational Diabetes: Systematic Review and Meta-Analysis. BMJ 2020, 369, m1361. [Google Scholar] [CrossRef]
- Li, Z.; Cheng, Y.; Wang, D.; Chen, H.; Chen, H.; Ming, W.-K.; Wang, Z. Incidence Rate of Type 2 Diabetes Mellitus after Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis of 170,139 Women. J. Diabetes Res. 2020, 2020, 3076463. [Google Scholar] [CrossRef]
- Nielsen, K.K.; Kapur, A.; Damm, P.; de Courten, M.; Bygbjerg, I.C. From Screening to Postpartum Follow-up—The Determinants and Barriers for Gestational Diabetes Mellitus (GDM) Services, a Systematic Review. BMC Pregnancy Childbirth 2014, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Herath, H.; Herath, R.; Wickremasinghe, R. Gestational Diabetes Mellitus and Risk of Type 2 Diabetes 10 Years after the Index Pregnancy in Sri Lankan Women—A Community Based Retrospective Cohort Study. PLoS ONE 2017, 12, e0179647. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Newton, K.M.; Knopp, R.H. Gestational Diabetes and the Incidence of Type 2 Diabetes: A Systematic Review. Diabetes Care 2002, 25, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Dennison, R.A.; Chen, E.S.; Green, M.E.; Legard, C.; Kotecha, D.; Farmer, G.; Sharp, S.J.; Ward, R.J.; Usher-Smith, J.A.; Griffin, S.J. The Absolute and Relative Risk of Type 2 Diabetes after Gestational Diabetes: A Systematic Review and Meta-Analysis of 129 Studies. Diabetes Res. Clin. Pract. 2021, 171, 108625. [Google Scholar] [CrossRef]
- Bellamy, L.; Casas, J.-P.; Hingorani, A.D.; Williams, D. Type 2 Diabetes Mellitus after Gestational Diabetes: A Systematic Review and Meta-Analysis. Lancet Lond. Engl. 2009, 373, 1773–1779. [Google Scholar] [CrossRef]
- Kramer, C.K.; Campbell, S.; Retnakaran, R. Gestational Diabetes and the Risk of Cardiovascular Disease in Women: A Systematic Review and Meta-Analysis. Diabetologia 2019, 62, 905–914. [Google Scholar] [CrossRef]
- Li, J.; Song, C.; Li, C.; Liu, P.; Sun, Z.; Yang, X. Increased Risk of Cardiovascular Disease in Women with Prior Gestational Diabetes: A Systematic Review and Meta-Analysis. Diabetes Res. Clin. Pract. 2018, 140, 324–338. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, S.; Sun, L.; Yang, H.; Jin, B.; Cao, X. Metabolic Syndrome Risk after Gestational Diabetes: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e87863. [Google Scholar] [CrossRef]
- Pathirana, M.M.; Lassi, Z.S.; Ali, A.; Arstall, M.A.; Roberts, C.T.; Andraweera, P.H. Association between Metabolic Syndrome and Gestational Diabetes Mellitus in Women and Their Children: A Systematic Review and Meta-Analysis. Endocrine 2021, 71, 310–320. [Google Scholar] [CrossRef]
- Aceti, A.; Santhakumaran, S.; Logan, K.M.; Philipps, L.H.; Prior, E.; Gale, C.; Hyde, M.J.; Modi, N. The Diabetic Pregnancy and Offspring Blood Pressure in Childhood: A Systematic Review and Meta-Analysis. Diabetologia 2012, 55, 3114–3127. [Google Scholar] [CrossRef]
- Philipps, L.H.; Santhakumaran, S.; Gale, C.; Prior, E.; Logan, K.M.; Hyde, M.J.; Modi, N. The Diabetic Pregnancy and Offspring BMI in Childhood: A Systematic Review and Meta-Analysis. Diabetologia 2011, 54, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Pathirana, M.M.; Lassi, Z.S.; Roberts, C.T.; Andraweera, P.H. Cardiovascular Risk Factors in Offspring Exposed to Gestational Diabetes Mellitus in Utero: Systematic Review and Meta-Analysis. J. Dev. Orig. Health Dis. 2020, 11, 599–616. [Google Scholar] [CrossRef]
- Abokaf, H.; Shoham-Vardi, I.; Sergienko, R.; Landau, D.; Sheiner, E. In Utero Exposure to Gestational Diabetes Mellitus and Long Term Endocrine Morbidity of the Offspring. Diabetes Res Clin Pr. 2018, 144, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Liu, E.; Qiao, Y.; Katzmarzyk, P.T.; Chaput, J.-P.; Fogelholm, M.; Johnson, W.D.; Kuriyan, R.; Kurpad, A.; Lambert, E.V.; et al. Maternal Gestational Diabetes and Childhood Obesity at Age 9–11: Results of a Multinational Study. Diabetologia 2016, 59, 2339–2348. [Google Scholar] [CrossRef]
- Holder, T.; Giannini, C.; Santoro, N.; Pierpont, B.; Shaw, M.; Duran, E.; Caprio, S.; Weiss, R. A Low Disposition Index in Adolescent Offspring of Mothers with Gestational Diabetes: A Risk Marker for the Development of Impaired Glucose Tolerance in Youth. Diabetologia 2014, 57, 2413–2420. [Google Scholar] [CrossRef]
- Clausen, T.D.; Mathiesen, E.R.; Hansen, T.; Pedersen, O.; Jensen, D.M.; Lauenborg, J.; Damm, P. High Prevalence of Type 2 Diabetes and Pre-Diabetes in Adult Offspring of Women with Gestational Diabetes Mellitus or Type 1 Diabetes: The Role of Intrauterine Hyperglycemia. Diabetes Care 2008, 31, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bacelis, J.; Sole-Navais, P.; Srivastava, A.; Juodakis, J.; Rouse, A.; Hallman, M.; Teramo, K.; Melbye, M.; Feenstra, B.; et al. Dissecting Maternal and Fetal Genetic Effects Underlying the Associations between Maternal Phenotypes, Birth Outcomes, and Adult Phenotypes: A Mendelian-Randomization and Haplotype-Based Genetic Score Analysis in 10,734 Mother–Infant Pairs. PLOS Med. 2020, 17, e1003305. [Google Scholar] [CrossRef] [PubMed]
- Solé-Navais, P.; Flatley, C.; Steinthorsdottir, V.; Vaudel, M.; Juodakis, J.; Chen, J.; Laisk, T.; LaBella, A.L.; Westergaard, D.; Bacelis, J.; et al. Genetic Effects on the Timing of Parturition and Links to Fetal Birth Weight. Nat. Genet. 2023, 55, 559–567. [Google Scholar] [CrossRef]
- Beaumont, R.N.; Flatley, C.; Vaudel, M.; Wu, X.; Chen, J.; Moen, G.-H.; Skotte, L.; Helgeland, Ø.; Solé-Navais, P.; Banasik, K.; et al. Genome-Wide Association Study of Placental Weight Identifies Distinct and Shared Genetic Influences between Placental and Fetal Growth. Nat. Genet. 2023, 55, 1807–1819. [Google Scholar] [CrossRef]
- Juliusdottir, T.; Steinthorsdottir, V.; Stefansdottir, L.; Sveinbjornsson, G.; Ivarsdottir, E.V.; Thorolfsdottir, R.B.; Sigurdsson, J.K.; Tragante, V.; Hjorleifsson, K.E.; Helgadottir, A.; et al. Distinction between the Effects of Parental and Fetal Genomes on Fetal Growth. Nat. Genet. 2021, 53, 1135–1142. [Google Scholar] [CrossRef]
- Beaumont, R.N.; Warrington, N.M.; Cavadino, A.; Tyrrell, J.; Nodzenski, M.; Horikoshi, M.; Geller, F.; Myhre, R.; Richmond, R.C.; Paternoster, L.; et al. Genome-Wide Association Study of Offspring Birth Weight in 86 577 Women Identifies Five Novel Loci and Highlights Maternal Genetic Effects That Are Independent of Fetal Genetics. Hum. Mol. Genet. 2018, 27, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Warrington, N.M.; Beaumont, R.N.; Horikoshi, M.; Day, F.R.; Helgeland, Ø.; Laurin, C.; Bacelis, J.; Peng, S.; Hao, K.; Feenstra, B.; et al. Maternal and Fetal Genetic Effects on Birth Weight and Their Relevance to Cardio-Metabolic Risk Factors. Nat. Genet. 2019, 51, 804–814. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).