

Lipid Metabolism and Statin Therapy in Neurodegenerative Diseases: An Endocrine View

 and
and
Abstract
1. Introduction
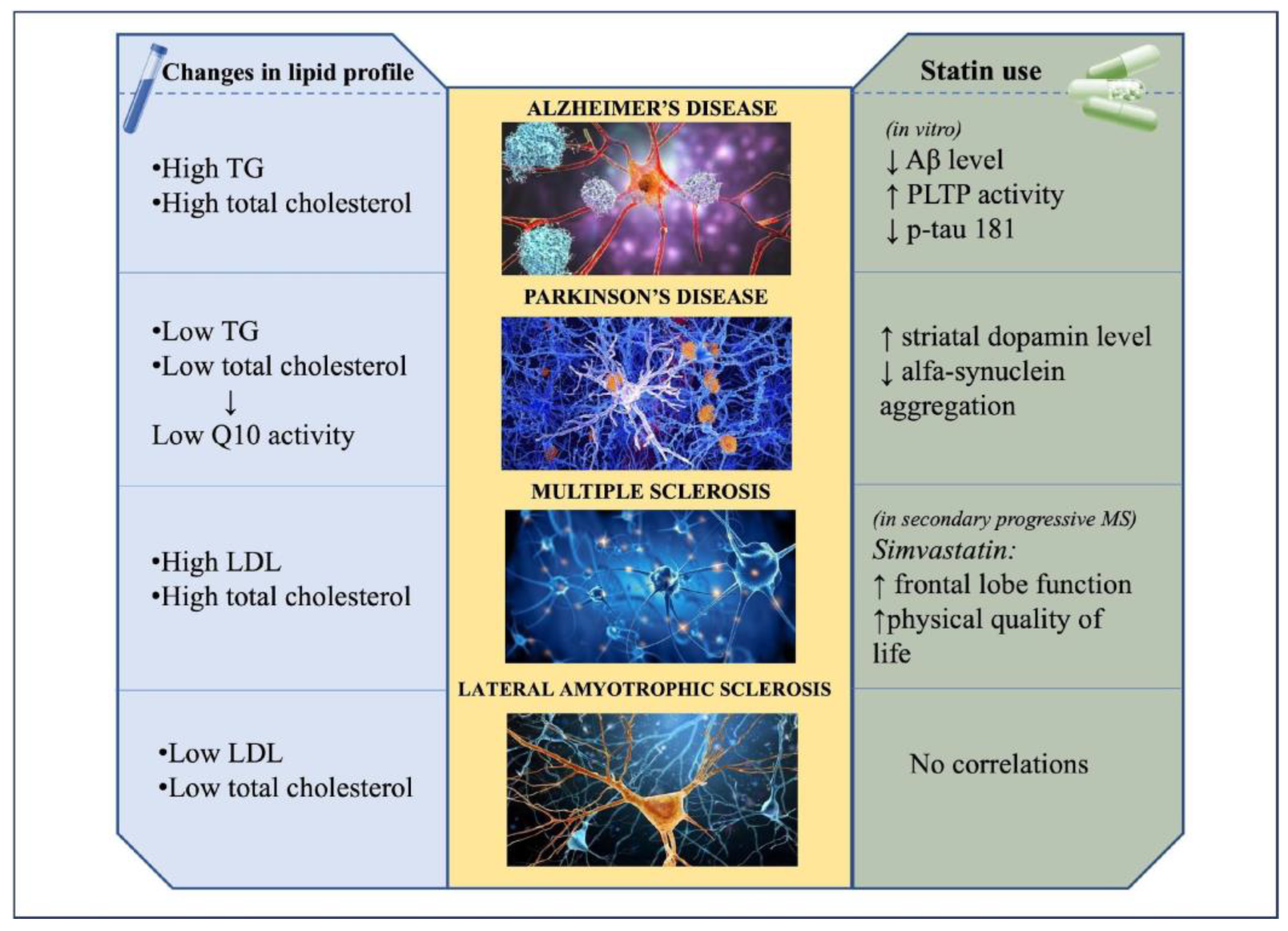
2. Alzheimer’s Disease
2.1. Lipids and Alzheimer’s Disease
2.2. Statin Use and Alzheimer’s Disease
3. Parkinson’s Disease
3.1. Lipids and Parkinson’s Disease
3.2. Statin Use and Risk of Parkinson’s Disease
4. Multiple Sclerosis
4.1. Lipids and Multiple Sclerosis
4.2. Statin Use and Risk of Multiple Sclerosis
5. Amyotrophic Lateral Sclerosis
5.1. Lipids and Amyotrophic Lateral Sclerosis
5.2. Statin Use and Amyotrophic Lateral Sclerosis

{kind=link}
| Authors’ Conclusions | Neurological Disease | Population of the Study | Type of Study | Aim of Study | Reference |
|---|---|---|---|---|---|
| The cohort taking statins during the study period had a 60% to 73% (p < 0.001) lower prevalence of probable AD. | Alzheimer’s Disease | 57,104 total patient from 3 different hospitals | Cross-sectional analysis | The aim of the study was to compare the prevalence of AD in patients 60 years or older in the following 3 groups: (1) the entire population; (2) patients receiving statins; (3) patients receiving medications used to treat hypertension or cardiovascular disease. | Wolozin B. et al., 2000 [56] |
| Treatment with atorvastatin could have a potential clinical benefit in patients with AD. | Alzheimer’s Disease | 63 participants with mild to moderate Alzheimer’s disease: -31 assigned to placebo -32 assigned to 80 mg atorvastatin time of observation: one year | Placebo-controlled randomized trial | The primary outcome measures were change in Alzheimer’s Disease Assessment Scale–cognitive subscale [ADAS-Cog] and the Clinical Global Impression of Change Scale scores [ADCS-CGIC]. | Sparks DL et al., 2005 [62] |
| Atorvastatin was not associated with significant clinical benefit over 72 weeks. However, patients enrolled did not need statin treatment, due to their good lipidic profile. | Alzheimer’s Disease | 640 participants with mild to moderate Alzheimer’s disease: -326 assigned to placebo -314 assigned to 80 mg atorvastatin/day Time of observation: 72 weeks followed by a 8-week atorvastatin withdrawal phase | Multicenter, double-blind, randomized | Coprimary endpoints were changes in cognition [ADAS-Cog] and global function [ADCS-CGIC]) at 72 weeks. | Feldman HH et al., 2010 [63] |
| Early statin use was significantly associated with a reduction in AD progression in mild-to-moderate AD patients. | Alzheimer’s Disease | 23.074 million people (total population of Taiwanese citizens seen in general medical practice) Time of observation: one year after starting use of any acetylcholinesterase inhibitors | Case-control study | Alzheimer disease patients with early statin use (before AChEI treatment) were those receiving any statin treatment during the exposure period. The primary outcome was the discontinuation of AChEI treatment, indicating AD progression. | Lin F-C et al., 2015 [65] |
| Statin use reduces the risk of PD. | Parkinson’s Disease | 1,457,836 participants without PD 15,102 participants with PD | Meta-analysis of: 5 case–control studies 3 cohort studies | The aim was to study the association between statin use and risk of developing PD. | Undela et al., 2013 [92] |
| High dose of Simvastatin had a positive effect on frontal lobe function. No other significant effects on the neurological outcome. | Multiple sclerosis | 140 patients with secondary progressive multiple sclerosis (SPMS): -70 assigned to placebo -70 assigned to 80 mg simvastatin Time of observation: 24th months | Clinical trial | The aim of the study was to investigate the effect of high-dose simvastatin on cognitive, neuropsychiatric, and health-related quality-of-life (HRQoL) outcome measure in patients with SPMS. Assessments were conducted at study entry, 12 months, and 24 months. | Chan et al., 2017 [107] |
| No definite association was found between statin use and the development of ALS. | Amyotrophic Lateral Sclerosis | 11,747 participants with ALS 230,573 participants without ALS | PRISMA meta-analisis: 3 case-control studies and 1 cohort study | The incidence of ALS in statin- and non-statin-treated patients was measured. The principal aim was to determine the effect of statins on ALS incidence. | Chang MC et al., 2021 [135] |
6. Genetic Mutations and Neurodegenerative Diseases: Correlation with Lipid Metabolism
7. Autophagy, Lysosomal Degradation, and Effects on Lipid Metabolism
8. Obesity and Neurodegenerative Diseases
9. Discussion and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Central nervous system | CNS |
| Genome-wide association studies | GWAS |
| Neurodegenerative diseases | NDDs |
| Alzheimer’s disease | AD |
| Parkinson’s disease | PD |
| Extracellular vesicles | EVs |
| Amyotrophic lateral sclerosis | ALS |
| Low density lipoprotein-cholesterol | LDL-C |
| Peripheral nervous system | PNS |
| Lipid-lowering molecules | LLMs |
| Amyloid-β | Aβ |
| Sphingolipids | SPs |
| Triglycerides | TGs |
| High density lipoprotein–cholesterol | HDL-C |
| Pittsburgh Compound B | PIB |
| Phospholipid transporter | PLTP |
| Neurofibrillary tangle | NFTs |
| Blood–brain barrier | BBB |
| Multiple sclerosis | MS |
| Body mass index | BMI |
| Glucocerebrosidase | GBA |
References
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Saher, G.; Quintes, S.; Nave, K.A. Cholesterol: A novel regulatory role in myelin formation. Neuroscientist 2011, 17, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Sooksawate, T.; Simmonds, M.A. Influence of membrane cholesterol on modulation of the GABA(A) receptor by neuroactive steroids and other potentiators. Br. J. Pharmacol. 2001, 134, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V. Lipids in Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 11523. [Google Scholar] [CrossRef]
- Pfrieger, F.W. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol. Life Sci. 2003, 60, 1158–1171. [Google Scholar] [CrossRef]
- Korade, Z.; Kenworthy, A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008, 55, 1265–1273. [Google Scholar] [CrossRef]
- Sebastiao, A.M.; Colino-Oliveira, M.; Assaife-Lopes, N.; Dias, R.B.; Ribeiro, J.A. Lipid rafts, synaptic transmission and plasticity: Impact in age-related neurodegenerative diseases. Neuropharmacology 2013, 64, 97–107. [Google Scholar] [CrossRef]
- Estes, R.E.; Lin, B.; Khera, A.; Davis, M.Y. Lipid Metabolism Influence on Neurodegenerative Disease Progression: Is the Vehicle as Important as the Cargo? Front. Mol. Neurosci. 2021, 14, 788695. [Google Scholar] [CrossRef]
- Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Zambón, D.; Quintana, M.; Mata, P.; Alonso, R.; Benavent, J.; Cruz-Sánchez, F.; Gich, J.; Pocoví, M.; Civeira, F.; Capurro, S.; et al. Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am. J. Med. 2010, 123, 267–274. [Google Scholar] [CrossRef]
- Gupta, S. Racial and ethnic disparities in subjective cognitive decline: A closer look, United States, 2015–2018. Gupta BMC Public Health 2021, 21, 1173. [Google Scholar] [CrossRef]
- Di Paolo, G.; Kim, T.-W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Gamba, P.; Testa, G.; Sottero, B.; Gargiulo, S.; Poli, G.; Leonarduzzi, G. The link between altered cholesterol metabolism and Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2012, 1259, 54–64. [Google Scholar] [CrossRef]
- Benn, M.; Nordestgaard, B.G.; Frikke-Schmidt, R.; Tybjærg-Hansen, A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer’s disease and Parkinson’s disease: Mendelian randomisation study. BMJ 2017, 357, j1648. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, J.; Zhao, K.; Gao, L.; Zhao, J. Cholesterol-induced toxicity: An integrated view of the role of cholesterol in multiple diseases. Cell Metab. 2021, 33, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Rajabian, A.; McCloskey, A.P.; Jamialahmadi, T.; Moallem, S.A.; Sahebkar, A. A review on the efficacy and safety of lipid-lowering drugs in neurodegenerative disease. Rev. Neurosci. 2023, 34, 801–824. [Google Scholar] [CrossRef]
- Rekatsina, M.; Paladini, A.; Piroli, A.; Zis, P.; Pergolizzi, J.V.; Varrassi, G. Pathophysiology and Therapeutic Perspectives of Oxidative Stress and Neurodegenerative Diseases: A Narrative Review. Adv. Ther. 2020, 37, 113–139. [Google Scholar] [CrossRef]
- Santa-Cecília, F.V.; Leite, C.A.; Del-Bel, E.; Raisman-Vozari, R. The Neuroprotective Effect of Doxycycline on Neurodegenerative Diseases. Neurotox. Res. 2019, 35, 981–986. [Google Scholar] [CrossRef]
- Dayar, E.; Pechanova, O. Targeted Strategy in Lipid-Lowering Therapy. Biomedicines 2022, 10, 1090. [Google Scholar] [CrossRef]
- Bird, T.D. Alzheimer Disease Overview. In GeneReviews®; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015. The Global Impact of Dementia. An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International (ADI): London, UK, 2015. [Google Scholar]
- Alzheimers Disease International; Guerchet, M.; Prince, M.; Prina, M. Numbers of People with Dementia Worldwide: An Update to the Estimates in the World Alzheimer Report 2015; Alzheimer’s Disease International: London, UK, 2020. [Google Scholar]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.R.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Solomon, A. Cholesterol as a risk factor for Alzheimer’s disease—Epidemiological evidence. Acta Neurol. Scand. 2006, 185, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Refolo, L.M.; Malester, B.; LaFrancois, J.; Bryant-Thomas, T.; Wang, R.; Tint, G.S.; Sambamurti, K.; Duff, K.; Pappolla, M.A. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000, 7, 321–331. [Google Scholar] [CrossRef]
- Shie, F.-S.; Jin, L.-W.; Cook, D.G.; Leverenz, J.B.; LeBoeuf, R.C. Diet-induced hypercholesterolemia enhances brain A beta accumulation in transgenic mice. Neuroreport 2002, 13, 455–459. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, X.; Shen, J.; Jiao, L.; Tong, J.; Zhao, W.; Du, K.; Gong, S.; Liu, M.; Wei, M. Elevated serum TC and LDL-C levels in Alzheimer’s disease and mild cognitive impairment: A meta-analysis study. Brain Res. 2020, 1727, 146554. [Google Scholar] [CrossRef]
- Nägga, K.; Gustavsson, A.-M.; Stomrud, E.; Lindqvist, D.; van Westen, D.; Blennow, K.; Zetterberg, H.; Melander, O.; Hansson, O. Increased midlife triglycerides predict brain β-amyloid and tau pathology 20 years later. Neurology 2018, 90, e73–e81. [Google Scholar] [CrossRef]
- Solomon, A.; Kåreholt, I.; Ngandu, T.; Winblad, B.; Nissinen, A.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Serum cholesterol changes after midlife and late-life cognition: Twenty-one-year follow-up study. Neurology 2007, 68, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Zandi, P.P.; Shao, H.; Waern, M.; Östling, S.; Guo, X.; Björkelund, C.; Lissner, L.; Skoog, I.; Gustafson, D.R. The 32-year relationship between cholesterol and dementia from midlife to late life. Neurology 2010, 75, 1888–1895. [Google Scholar] [CrossRef]
- Mielke, M.M.; Zandi, P.P.; Sjögren, M.; Gustafson, D.; Ostling, S.; Steen, B.; Skoog, I. High totaln cholesterol levels in late life associated with a reduced risk of dementia. Neurology 2005, 64, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Shofer, J.B.; Kukull, W.A.; Peskind, E.R.; Tsuang, D.W.; Breitner, J.C.S.; McCormick, W.; Bowen, J.D.; Teri, L.; Schellenberg, G.D.; et al. Serum cholesterol and risk of Alzheimer disease: A community-based cohort study. Neurology 2005, 65, 1045–1050. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Bryant-Thomas, T.K.; Herbert, D.; Pacheco, J.; Fabra Garcia, M.; Manjon, M.; Girones, X.; Henry, T.L.; Matsubara, E.; Zambon, D.; et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 2003, 61, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Bettcher, B.M.; Ard, M.C.; Reed, B.R.; Benitez, A.; Simmons, A.; Larson, E.B.; Sonnen, J.A.; Montine, T.J.; Li, G.; Keene, C.D.; et al. Association between cholesterol exposure and neuropathological findings: The ACT Study. J. Alzheimers Dis. 2017, 59, 1307–1315. [Google Scholar] [CrossRef]
- Giussani, P.; Prinetti, A.; Tringali, C. The Role of Sphingolipids in Myelination and Myelin Stability and Their Involvement in Childhood and Adult Demyelinating Disorders. J. Neurochem. 2021, 156, 403–414. [Google Scholar] [CrossRef]
- Varma, V.R.; Oommen, A.M.; Varma, S.; Casanova, R.; An, Y.; Andrews, R.M.; O’Brien, R.; Pletnikova, O.; Troncoso, J.C.; Toledo, J.; et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med. 2018, 15, e1002482. [Google Scholar] [CrossRef] [PubMed]
- Alessenko, A.V.; Bugrova, A.E.; Dudnik, L.B. Connection of lipid peroxide oxidation with the sphingomyelin pathway in the development of Alzheimer’s disease. Biochem. Soc. Trans. 2004, 32 Pt 1, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Giovagnoni, C.; Ali, M.; Eijssen, L.M.T.; Maes, R.; Choe, K.; Mulder, M.; Kleinjans, J.; del Sol, A.; Glaab, E.; Mastroeni, D.; et al. Altered Sphingolipid Function in Alzheimer’s Disease; a Gene Regulatory Network Approach. Neurobiol. Aging 2021, 102, 178–187. [Google Scholar] [CrossRef]
- Katsel, P.; Li, C.; Haroutunian, V. Gene Expression Alterations in the Sphingolipid Metabolism Pathways during Progression of Dementia and Alzheimer’s Disease: A Shift Toward Ceramide Accumulation at the Earliest Recognizable Stages of Alzheimer’s Disease? Neurochem. Res. 2007, 32, 845–856. [Google Scholar] [CrossRef]
- Burgess, B.L.; McIsaac, S.A.; Naus, K.E.; Chan, J.Y.; Tansley, G.H.; Yang, J.; Miao, F.; Ross, C.; Van Eck, M.; Hayden, M. Elevated plasma triglyceride levels precede amyloid deposition in Alzheimer’s disease mouse models with abundant Aβ in plasma. Neurobiol. Dis. 2006, 24, 114–127. [Google Scholar] [CrossRef]
- Amelianchik, A.; Sweetland-Martin, L.; Norris, E.H. The effect of dietary fat consumption on Alzheimer’s disease pathogenesis in mouse models. Transl. Psychiatry 2022, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Byun, M.S.; Yi, D.; Choe, Y.M.; Sohn, B.K.; Baek, H.W.; Lee, J.H.; Kim, H.J.; Han, J.Y.; Yoon, E.J.; et al. Association between serum triglycerides and cerebral amyloidosis in cognitively normal elderly. Am. J. Geriatr. Psychiatry 2016, 24, 604–612. [Google Scholar]
- Peloso, G.M.; Beiser, A.S.; DeStefano, A.L.; Seshadri, S. Genetic interaction with plasma lipids on Alzheimer’s disease in the Framingham heart study. J. Alzheimer’s Dis. 2018, 66, 1275–1282. [Google Scholar]
- Reed, B.; Villeneuve, S.; Mack, W.; DeCarli, C.; Chui, H.C.; Jagust, W. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 2014, 71, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimer’s Dement. 2017, 13, 140–151. [Google Scholar] [CrossRef]
- Lepara, O.; Valjevac, A.; Alajbegović, A.; Zaćiragić, A.; Nakaš-Ićindić, E. Decreased serum lipids in patients with probable Alzheimer’s disease. Bosn. J. Basic. Med. Sci. 2009, 9, 215–220. [Google Scholar] [CrossRef]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar]
- Kurata, T.; Miyazaki, K.; Kozuki, M.; Morimoto, N.; Ohta, Y.; Ikeda, Y.; Abe, K. Atorvastatin and pitavastatin reduce senile plaques and inflammatory responses in a mouse model of Alzheimer’s disease. Neurol. Res. 2012, 34, 601–610. [Google Scholar] [CrossRef]
- Li, L.; Cao, D.; Kim, H.; Lester, R.; Fukuchi, K.-I. Simvastatin enhances learning and memory independent of amyloid load in mice. Ann. Neurol. 2006, 60, 729–739. [Google Scholar]
- Tong, X.-K.; Lecrux, C.; Rosa-Neto, P.; Hamel, E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. J. Neurosci. 2012, 32, 4705–4715. [Google Scholar] [CrossRef]
- Boimel, M.; Grigoriadis, N.; Lourbopoulos, A.; Touloumi, O.; Rosenmann, D.; Abramsky, O.; Rosenmann, H. Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. J. Neuropathol. Exp. Neurol. 2009, 68, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Kirkland, S.; Hogan, D.B.; MacKnight, C.; Merry, H.; Verreault, R.; Wolfson, C.; McDowell, I. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch. Neurol. 2002, 59, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Rea, T.D.; Breitner, J.C.; Psaty, B.M.; Fitzpatrick, A.L.; Lopez, O.L.; Newman, A.B.; Hazzard, W.R.; Zandi, P.P.; Burke, G.L.; Lyketsos, C.G.; et al. Statin use and the risk of incident dementia: The Cardiovascular Health Study. Arch. Neurol. 2005, 62, 1047–1051. [Google Scholar] [CrossRef]
- Cramer, C.; Haan, M.N.; Galea, S.; Langa, K.M.; Kalbfleisch, J.D. Use of statins and incidence of dementia and cognitive impairment without dementia in a cohort study. Neurology 2008, 71, 344–350. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef]
- Sparks, D.L.; Sabbagh, M.N.; Connor, D.J.; Lopez, J.; Launer, L.J.; Browne, P.; Wasser, D.; Johnson-Traver, S.; Lochhead, J.; Ziolwolski, C. Atorvastatin for the treatment of mild to moderate Alzheimer disease: Preliminary results. Arch. Neurol. 2005, 62, 753–757. [Google Scholar] [CrossRef]
- Feldman, H.H.; Doody, R.S.; Kivipelto, M.; Sparks, D.L.; Waters, D.D.; Jones, R.W.; Schwam, E.; Schindler, R.; Hey-Hadavi, J.; LEADe Investigators. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology 2010, 74, 956–964. [Google Scholar] [CrossRef]
- Sano, M.; Bell, K.L.; Galasko, D.; Galvin, J.E.; Thomas, R.G.; van Dyck, C.H.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 2011, 77, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.C.; Chuang, Y.S.; Hsieh, H.M.; Lee, T.C.; Chiu, K.F.; Liu, C.K.; Wu, M.T. Early statin use and the progression of Alzheimer disease. Medicine 2015, 94, 47. [Google Scholar] [CrossRef] [PubMed]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-Wide Association Study Reveals Genetic Risk Underlying Parkinson’s Disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Galper, J.; Dean, N.J.; Pickford, R.; Lewis, S.J.G.; Halliday, G.M.; Kim, W.S.; Dzamko, N. Lipid Pathway Dysfunction Is Prevalent in Patients with Parkinson’s Disease. Brain 2022, 145, 3472–3487. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s Disease: An Introspection of Its Journey towards Precision Medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Blandini, F.; Cilia, R.; Cerri, S.; Pezzoli, G.; Schapira, A.H.V.; Mullin, S.; Lanciego, J.L. Glucocerebrosidase Mutations and Synucleinopathies: Toward a Model of Precision Medicine. Mov. Disord. 2019, 34, 9–21. [Google Scholar] [CrossRef]
- Fais, M.; Dore, A.; Galioto, M.; Galleri, G.; Crosio, C.; Iaccarino, C. Parkinson’s Disease-Related Genes and Lipid Alteration. Int. J. Mol. Sci. 2021, 22, 7630. [Google Scholar] [CrossRef]
- Lansbury, P. The sphingolipids clearly play a role in Parkinson’s disease, but nature has made it complicated. Mov. Disord. 2022, 37, 1985–1989. [Google Scholar] [CrossRef]
- Lin, G.; Wang, L.; Marcogliese, P.C.; Bellen, H.J. Sphingolipids in the pathogenesis of Parkinson’s disease and parkinsonism. Trends Endocrinol. Metab. 2019, 30, 106–117. [Google Scholar] [CrossRef]
- Yang, R.; He, C.; Zhang, P.; Li, Y.; Rong, S.; Chen, X.; Qi, Q.; Gao, Z.; Chi, J.; Wang, L.; et al. Plasma sphingolipids, dopaminergic degeneration and clinical progression in idiopathic Parkinson’s disease. Park. Relat. Disord. 2024, 126, 107071. [Google Scholar] [CrossRef] [PubMed]
- Abbott, S.K.; Li, H.; Munoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease: Altered Ceramide in Parkinson’s Disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef]
- Galvagnion, C.; Marlet, F.R.; Cerri, S.; Schapira, A.H.V.; Blandini, F.; Di Monte, D.A. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. Brain 2022, 145, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Huebecker, M.; Moloney, E.B.; Van Der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef]
- Galper, J.; Kim, W.S.; Dzamko, N. LRRK2 and lipid pathways: Implications for Parkinson’s disease. Biomolecules 2022, 12, 1597. [Google Scholar] [CrossRef]
- Signorelli, P.; Conte, C.; Albi, E. The multiple roles of sphingomyelin in Parkinson’s disease. Biomolecules 2021, 11, 1311. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Barrantes, F.J. Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 2345–2361. [Google Scholar] [CrossRef]
- Won, J.H.; Kim, S.K.; Shin, I.C.; Ha, H.C.; Jang, J.M.; Back, M.J.; Kim, D.K. Dopamine transporter trafficking is regulated by neutral sphingomyelinase 2/ceramide kinase. Cell. Signal. 2018, 44, 171–187. [Google Scholar] [CrossRef]
- Fu, X.; Wang, Y.; He, X.; Li, H.; Liu, H.; Zhang, X. A Systematic Review and Meta-Analysis of Serum Cholesterol and Triglyceride Levels in Patients with Parkinson’s Disease. Lipids Health Dis. 2020, 19, 97. [Google Scholar] [CrossRef]
- Fang, F.; Zhan, Y.; Hammar, N.; Shen, X.; Wirdefeldt, K.; Walldius, G.; Mariosa, D. Lipids, Apolipoproteins, and the Risk of Parkinson Disease: A Prospective Cohort Study and a Mendelian Randomization Analysis. Circ. Res. 2019, 125, 643–652. [Google Scholar] [CrossRef]
- Kaikkonen, J.; Nyyssönen, K.; Tuomainen, T.-P.; Ristonmaa, U.; Salonen, J.T. Determinants of Plasma Coenzyme Q 10 in Humans. FEBS Lett. 1999, 443, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Kabuto, H.; Yamanushi, T.T.; Janjua, N.; Takayama, F.; Mankura, M. Effects of Squalene/Squalane on Dopamine Levels, Antioxidant Enzyme Activity, and Fatty Acid Composition in the Striatum of Parkinson’s Disease Mouse Model. J. Oleo Sci. 2013, 62, 21–28. [Google Scholar] [CrossRef]
- Roy, A.; Ghosh, A.; Jana, A.; Liu, X.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Sodium Phenylbutyrate Controls Neuroinflammatory and Antioxidant Activities and Protects Dopaminergic Neurons in Mouse Models of Parkinson’s Disease. PLoS ONE 2012, 7, e38113. [Google Scholar] [CrossRef] [PubMed]
- Gaig, C.; Tolosa, E. When Does Parkinson’s Disease Begin? Mov. Disord. 2009, 24, S656–S664. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hasegawa, M.; Takano, O. The effect of dopamine on serum lipid concentration after propofol administration. Masui. Jpn. J. Anesthesiol. 2002, 51, 286–288. [Google Scholar]
- Scigliano, G.; Musicco, M.; Soliveri, P.; Piccolo, I.; Ronchetti, G.; Girotti, F. Reduced Risk Factors for Vascular Disorders in Parkinson Disease Patients: A Case-Control Study. Stroke 2006, 37, 1184–1188. [Google Scholar] [CrossRef]
- Scigliano, G.; Ronchetti, G.; Girotti, F.; Musicco, M. Sympathetic Modulation by Levodopa Reduces Vascular Risk Factors in Parkinson Disease. Park. Relat. Disord. 2009, 15, 138–143. [Google Scholar] [CrossRef]
- Sheng, Z.; Jia, X.; Kang, M. Statin use and risk of Parkinson’s disease: A meta-analysis. Behav. Brain Res. 2016, 309, 29–34. [Google Scholar] [CrossRef]
- Undela, K.; Gudala, K.; Malla, S.; Bansal, D. Statin Use and Risk of Parkinson’s Disease: A Meta-Analysis of Observational Studies. J. Neurol. 2013, 260, 158–165. [Google Scholar] [CrossRef]
- Wood, W.G.; Eckert, G.P.; Igbavboa, U.; Müller, W.E. Statins and Neuroprotection: A Prescription to Move the Field Forward. Ann. N. Y. Acad. Sci. 2010, 1199, 69–76. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, J.; Chen, X.; Li, J.; Yang, Y.; Weng, J.; Deng, C.; Yenari, M.A. Statins: Multiple Neuroprotective Mechanisms in Neurodegenerative Diseases. Exp. Neurol. 2011, 230, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L. Simvastatin Prevents 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Striatal Dopamine Depletion and Protein Tyrosine Nitration in Mice. Brain Res. 2005, 1037, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rajanikant, G.; Zemke, D.; Kassab, M.; Majid, A. The Therapeutic Potential of Statins in Neurological Disorders. Curr. Med. Chem. 2007, 14, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Jakimovski, D.; Bittner, S.; Zivadinov, R.; Morrow, S.A.; Benedict, R.H.; Zipp, F.; Weinstock-Guttman, B. Multiple sclerosis. Lancet 2024, 403, 183–202. [Google Scholar] [CrossRef]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers. 2018, 4, 43. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef]
- Wekerle, H.; Lassmann, H. The immunology of multiple sclerosis. In McAlpine’s Multiple Sclerosis; Compston, A., Ed.; Churchill Livingstone: London, UK, 1998; pp. 379–407. [Google Scholar]
- Steinmetz, A.; Hocke, G.; Saïle, R.; Puchois, P.; Fruchart, J.C. Influence of serum amyloid A on cholesterol esterification in human plasma. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1989, 1006, 173–178. [Google Scholar] [CrossRef]
- Ristori, G.; Laurenti, F.; Stacchini, P.; Gasperini, C.; Buttinelli, C.; Pozzilli, C.; Salvetti, M. Serum amyloid A protein is elevated in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 1998, 88, 9–12. [Google Scholar] [CrossRef]
- Giubilei, F.; Antonini, G.; Di Legge, S.; Sormani, M.P.; Pantano, P.; Antonini, R.; Sepe-Monti, M.; Caramia, F.; Pozzilli, C. Blood cholesterol and MRI activity in first clinical episode suggestive of multiple sclerosis. Acta Neurol. Scand. 2002, 106, 109–112. [Google Scholar] [CrossRef]
- Newcombe, J.; Li, H.; Cuzner, M.L. Low density lipoprotein uptake by macrophages in multiple sclerosis plaques:implications for pathogenesis. Neuropathol. Appl. Neurobiol. 1994, 20, 152–162. [Google Scholar] [CrossRef]
- Stampanoni Bassi, M.; Iezzi, E.; Buttari, F.; Gilio, L.; Simonelli, I.; Carbone, F.; Micillo, T.; De Rosa, V.; Sica, F.; Furlan, R.; et al. Obesity worsens central inflammation and disability in multiple sclerosis. Mult. Scler. J. 2020, 26, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Noori, H.; Gheini, M.R.; Rezaeimanesh, N.; Saeedi, R.; Rezaei Aliabadi, H.; Sahraian, M.A.; Naser Moghadasi, A. The correlation between dyslipidemia and cognitive impairment in multiple sclerosis patients. Mult. Scler. Relat. Disord. 2019, 36, 101415. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Binks, S.; Nicholas, J.M.; Frost, C.; Cardoso, M.J.; Ourselin, S.; Wilkie, D.; Nicholas, R.; Chataway, J. Effect of high-dose simvastatin on cognitive, neuropsychiatric, and health-related quality-of-life measures in secondary progressive multiple sclerosis: Secondary analyses from the MS-STAT randomised, placebo-controlled trial. Lancet Neurol. 2017, 16, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Tettey, P.; Simpson, S., Jr.; Taylor, B.; Blizzard, L.; Ponsonby, A.L.; Dwyer, T.; Kostner, K.; van der Mei, I. An adverse lipid profile is associated with disability and progression in disability, in people with MS. Mult. Scler. 2014, 20, 1737–1744. [Google Scholar] [CrossRef]
- Gafson, A.R.; Thorne, T.; McKechnie, C.I.J.; Jimenez, B.; Nicholas, R.; Matthews, P.M. Lipoprotein markers associated with disability from multiple sclerosis. Sci. Rep. 2018, 8, 17026. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Saad, H.M.; Batiha, G.E.S. The potential therapeutic effect of statins in multiple sclerosis: Beneficial or detrimental effects. Inflammopharmacology 2023, 31, 1671–1682. [Google Scholar] [CrossRef]
- Batoee, S.; Etminaniesfahani, M.; Mazdeh, M.; Soltanian, A.; Nouri, F. Evaluation of Rosuvastatin Therapy on SIRT1 Gene Expression in Patients with Multiple Sclerosis: An Uncontrolled Clinical Trial. Curr. Ther. Res. 2023, 99, 100718. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Search Results for “Multiple Sclerosis” and “Statins”. Available online: https://clinicaltrials.gov/search?cond=Multiple%20Sclerosis&term=statins (accessed on 1 April 2025).
- Blackstone, J.; Williams, T.; Nicholas, J.M.; Bordea, E.; Angelis, F.D.; Bianchi, A.; Calvi, A.; Doshi, A.; John, N.; Mangion, S.A.; et al. Evaluating the effectiveness of simvastatin in slowing the progression of disability in secondary progressive multiple sclerosis (MS-STAT2): Protocol for a multicentre, randomised controlled, double-blind, phase 3 clinical trial in the UK. BMJ Open 2024, 14, e086414. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef]
- Ghasemi, M.; Brown, R.H. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125. [Google Scholar] [CrossRef]
- Van Damme, P.; Dewil, M.; Robberecht, W.; Van Den Bosch, L. Excitotoxicity and Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2005, 2, 147–159. [Google Scholar] [PubMed]
- Greco, V.; Longone, P.; Spalloni, A.; Pieroni, L.; Urbani, A. Crosstalk between oxidative stress and mitochondrial damage: Focus on amyotrophic lateral sclerosis. In Mitochondria in Health and in Sickness; Urbani, A., Babu, M., Eds.; Springer: Singapore, 2019; pp. 71–82. ISBN 9789811383670. [Google Scholar]
- McCombe, A.P.; Henderson, D.R. The Role of Immune and Inflammatory Mechanisms in ALS. Curr. Mol. Med. 2011, 999, 246–254. [Google Scholar] [CrossRef]
- Agrawal, I.; Lim, Y.S.; Ng, S.Y.; Ling, S.C. Deciphering lipid dysregulation in ALS: From mechanisms to translational medicine. Transl. Neurodegener. 2022, 11, 48. [Google Scholar] [CrossRef]
- Phan, K.; He, Y.; Bhatia, S.; Pickford, R.; McDonald, G.; Mazumder, S.; Timmins, H.C.; Hodges, J.R.; Piguet, O.; Dzamko, N.; et al. Multiple pathways of lipid dysregulation in amyotrophic lateral sclerosis. Brain Commun. 2022, 5, fcac340. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.C.; Yu, J.; Sardi, S.P.; Lamya, S. Sterol auto-oxidation adversely affects human motor neuron viability and is a neuropathological feature of amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 803. [Google Scholar] [CrossRef]
- Michels, S.; Kurz, D.; Rosenbohm, A.; Peter, R.S.; Just, S.; Bäzner, H.; Börtlein, A.; Dettmers, C.; Gold, H.J.; ALS Registry Swabia Study Group; et al. Association of blood lipids with onset and prognosis of amyotrophic lateral sclerosis: Results from the ALS Swabia registry. J. Neurol. 2023, 270, 3082–3090. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Guo, X.; Chen, X.; Zheng, Z.; Wei, Q.; Cao, B.; Zeng, Y.; Shang, H. The Serum Lipid Profiles of Amyotrophic Lateral Sclerosis Patients: A Study from South-West China and a Meta-Analysis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 359–365. [Google Scholar] [CrossRef]
- Liu, J.; Luo, X.; Chen, X.; Shang, H. Lipid profile in patients with amyotrophic lateral sclerosis: A systematic review and meta-analysis. Front. Neurol. 2020, 11, 567753. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Acimovic, J.; Lövgren-Sandblom, A.; Parini, P.; Andersen, P.M.; Björkhem, I. Cholesterol, Oxysterol, Triglyceride, and Coenzyme Q Homeostasis in ALS. Evidence against the Hypothesis That Elevated 27-Hydroxycholesterol Is a Pathogenic Factor. PLoS ONE 2014, 9, e113619. [Google Scholar] [CrossRef]
- Dorst, J.; Kühnlein, P.; Hendrich, C.; Kassubek, J.; Sperfeld, A.D.; Ludolph, A.C. Patients with Elevated Triglyceride and Cholesterol Serum Levels Have a Prolonged Survival in Amyotrophic Lateral Sclerosis. J. Neurol. 2011, 258, 613–617. [Google Scholar] [CrossRef]
- Vejux, A.; Namsi, A.; Nury, T.; Moreau, T.; Lizard, G. Biomarkers of Amyotrophic Lateral Sclerosis: Current Status and Interest of Oxysterols and Phytosterols. Front. Mol. Neurosci. 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; De Aguilar, J.-L.G.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- Nakamura, R.; Kurihara, M.; Ogawa, N.; Kitamura, A.; Yamakawa, I.; Bamba, S.; Sanada, M.; Sasaki, M.; Urushitani, M. Investigation of the prognostic predictive value of serum lipid profiles in amyotrophic lateral sclerosis: Roles of sex and hypermetabolism. Sci. Rep. 2022, 12, 1826. [Google Scholar] [CrossRef]
- Ingre, C.; Chen, L.; Zhan, Y.; Termorshuizen, J.; Yin, L.; Fang, F. Lipids, apolipoproteins, and prognosis of amyotrophic lateral sclerosis. Neurology 2020, 94, e1835–e1844. [Google Scholar] [CrossRef]
- Janse van Mantgem, M.R.; Van Rheenen, W.; Hackeng, A.V.; Van Es, M.A.; Veldink, J.H.; Van Den Berg, L.H.; van Eijk, R.P. Association Between Serum Lipids and Survival in Patients with Amyotrophic Lateral Sclerosis A Meta-analysis and Population-Based Study. Neurology 2023, 100, e1062–e1071. [Google Scholar] [CrossRef]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Croixmarie, V.; Priestman, D.A.; Rosenbohm, A.; Dirrig-Grosch, S.; D’Ambra, E.; Huebecker, M.; Hussain, G.; Boursier-Neyret, C.; Echaniz-Laguna, A.; et al. Amyotrophic lateral sclerosis and denervation alter sphingolipids and up-regulate glucosylceramide synthase. Hum. Mol. Genet. 2015, 24, 7390–7405. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.C.; Treleaven, C.M.; Pacheco, J.; Cooper, S.; Bao, C.; Abraham, M.; Cromwell, M.; Sardi, S.P.; Chuang, W.L.; Sidman, R.L.; et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 8100–8105. [Google Scholar] [CrossRef]
- Chang, M.C.; Kwak, S.G.; Park, J.S.; Park, D. Relationship between statins and the risk of amyotrophic lateral sclerosis. Medicine 2021, 100, e26751. [Google Scholar] [CrossRef]
- Freedman, D.M.; Kuncl, R.W.; Cahoon, E.K.; Rivera, D.R.; Pfeiffer, R.M. Relationship of Statins and Other Cholesterol-Lowering Medications and Risk of Amyotrophic Lateral Sclerosis in the US Elderly. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 538–546. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.; Xia, K.; Huang, T.; Fan, D. Mendelian Randomization Analysis Reveals Statins Potentially Increase Amyotrophic Lateral Sclerosis Risk Independent of Peripheral Cholesterol-Lowering Effects. Biomedicine 2023, 11, 1359. [Google Scholar] [CrossRef] [PubMed]
- Zinman, L.; Sadeghi, R.; Gawel, M.; Patton, D.; Kiss, A. Are Statin Medications Safe in Patients with ALS? Amyotroph. Lateral Scler. 2008, 9, 223–228. [Google Scholar] [CrossRef]
- Vaage, A.M.; Holmøy, T.; Dahl, J.; Stigum, H.; Meyer, H.E.; Nakken, O. Statin Use and Amyotrophic Lateral Sclerosis Survival: A Population-Based Cohort Study. Eur. J. Neurol. 2025, 32, e70095. [Google Scholar] [CrossRef]
- Schumacher, J.; Peter, R.S.; Nagel, G.; Rothenbacher, D.; Rosenbohm, A.; Ludolph, A.C.; Dorst, J.; ALS Registry Swabia Study Group. Statins, Diabetes Mellitus and Prognosis of Amyotrophic Lateral Sclerosis: Data From 501 Patients of a Population-Based Registry in Southwest Germany. Eur. J. Neurol. 2020, 27, 1405–1414. [Google Scholar] [CrossRef]
- Drory, V.E.; Bronipolsky, T.; Artamonov, I.; Nefussy, B. Influence of Statins Treatment on Survival in Patients With Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2008, 273, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Kamano, S.; Ozawa, D.; Ikenaka, K.; Nagai, Y. Role of Lipids in the Pathogenesis of Parkinson’s disease. Int. J. Mol. Sci. 2024, 25, 8935. [Google Scholar] [CrossRef]
- Jellinger, K.A. Behavioral disorders in dementia with Lewy bodies: Old and new knowledge. J. Neural Transm. 2025, 132, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Giasson, B.I.; Chakrabarty, P. α-Synuclein and astrocytes: Tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. 2019, 138, 1–21. [Google Scholar] [CrossRef]
- Castagnet, P.I.; Golovko, M.Y.; Barceló-Coblijn, G.C.; Nussbaum, R.L.; Murphy, E.J. Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. J. Neurochem. 2005, 94, 839–849. [Google Scholar] [CrossRef]
- Galvagnion, C. The role of lipids interacting with α-synuclein in the pathogenesis of Parkinson’s disease. J. Park. Dis. 2017, 7, 433–450. [Google Scholar] [CrossRef]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Fujikake, N.; Takeuchi, T.; Kohyama-Koganeya, A.; Nakajima, K.; Hirabayashi, Y.; Wada, K.; Nagai, Y. Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant α-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 6675–6686. [Google Scholar] [CrossRef] [PubMed]
- Choong, C.J.; Aguirre, C.; Kakuda, K.; Beck, G.; Nakanishi, H.; Kimura, Y.; Shimma, S.; Nabekura, K.; Hideshima, M.; Doi, J.; et al. Phosphatidylinositol-3,4,5-trisphosphate interacts with alpha-synuclein and initiates its aggregation and formation of Parkinson’s disease-related fibril polymorphism. Acta Neuropathol. 2023, 145, 573–595. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. α-synuclein-harboring familial Parkinson’s disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in α synuclein mice: Implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef]
- Sapaly, D.; Cheguillaume, F.; Weill, L.; Clerc, Z.; Biondi, O.; Bendris, S.; Buon, C.; Slika, R.; Piller, E.; Sundaram, V.K.; et al. Dysregulation of muscle cholesterol transport in amyotrophic lateral sclerosis. Brain 2025, 148, 788–802. [Google Scholar] [CrossRef]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The role of skeletal muscle in amyotrophic lateral sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.L.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.P.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Gonzalez De Aguilar, J.L. Lipid biomarkers for amyotrophic lateral sclerosis. Front. Neurol. 2019, 10, 284. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, M.K.; Lee, E.; Bradburn, M.; McDermott, C.J.; Shaw, P.J. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: Insights from the olesoxime clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 478–484. [Google Scholar] [CrossRef]
- Seelen, M.; van Doormaal, P.T.; Visser, A.E.; Huisman, M.H.; Roozekrans, M.H.; de Jong, S.W.; van der Kooi, A.J.; de Visser, M.; Voermans, N.C.; Veldink, J.H.; et al. Prior medical conditions and the risk of amyotrophic lateral sclerosis. J. Neurol. 2014, 261, 1949–1956. [Google Scholar] [CrossRef]
- Mariosa, D.; Hammar, N.; Malmstrom, H.; Ingre, C.; Jungner, I.; Ye, W.; Fang, F.; Walldius, G. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: A more than 20-year follow-up of the Swedish AMORIS cohort. Ann. Neurol. 2017, 81, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X. Causal effects of blood lipids on amyotrophic lateral sclerosis: A Mendelian randomization study. Hum. Mol. Genet. 2019, 28, 688–697. [Google Scholar]
- Chen, X.; Yazdani, S.; Piehl, F.; Magnusson, P.K.E.; Fang, F. Polygenic link between blood lipids and amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 67, 202.e1–202.e6. [Google Scholar] [CrossRef] [PubMed]
- Bandres-Ciga, S.; Noyce, A.J.; Hemani, G.; Nicolas, A.; Calvo, A.; Mora, G.; ITALSGEN Consortium; International ALS Genomics Consortium; Tienari, P.J.; Stone, D.J.; et al. Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann. Neurol. 2019, 85, 470–481. [Google Scholar] [CrossRef]
- Nogueira-Machado, J.A.; Lima e Silva, F.D.C.; Rocha-Silva, F.; Gomes, N. Amyotrophic Lateral Sclerosis (ALS): An overview of genetic and metabolic signaling mechanisms. CNS Neurol. Disord. Drug Target. 2025, 24, 83–90. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar]
- Krishnamurthy, H.K.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Changalath, C.; Rajasekaran, J.J. An overview of the genes and biomarkers in Alzheimer’s disease. Ageing Res. Rev. 2025, 104, 102599. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [PubMed]
- Quaid, K.A. Genetic Counseling for Frontotemporal Dementias. J. Mol. Neurosci. 2011, 45, 706–709. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger TL, S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Dominantly Inherited Alzheimer Network Clinical biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Van Deerlin, V.M. The genetics and neuropathology of neurodegenerative disorders: Perspectives and implications for research and clinical practice. Acta Neuropathol. 2012, 124, 297–303. [Google Scholar] [CrossRef]
- Sawcer, S.; Maranian, M.; Setakis, E.; Curwen, V.; Akesson, E.; Hensiek, A.; Coraddu, F.; Roxburgh, R.; Sawcer, D.; Gray, J.; et al. A whole genome screen for linkage disequilibrium in multiple sclerosis confirms disease associations with regions previously linked to susceptibility. Brain 2002, 125, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 723–1737. [Google Scholar]
- Islam, S.; Noorani, A.; Sun, Y.; Michikawa, M.; Zou, K. Multi-functional role of apolipoprotein E in neurodegenerative diseases. Front. Aging Neurosci. 2025, 17, 1535280. [Google Scholar] [CrossRef]
- Puri, C.; Gratian, M.J.; Rubinsztein, D.C. Mammalian autophagosomes form from finger-like phagophores. Dev. Cell 2023, 58, 2746–2760. [Google Scholar] [CrossRef]
- Palmer, J.E.; Wilson, N.; Son, S.M.; Obrocki, P.; Wrobel, L.; Rob, M.; Takla, M.; Korolchuk, V.I. Rubinsztein DCAutophagy, aging, and age-related neurodegeneration. Neuron 2025, 113, 29–48. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Jaishy, B.; Abel, E.D. Lipids, lysosomes, and autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef] [PubMed]
- Seungyoon, B.Y.; Pekkurnaz, G. Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J. Mol. Biol. 2018, 4320, 3922–3941. [Google Scholar]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Wang, L.; Gao, Y.; Feng, G.; Li, G.; Zou, J.; Yu, M.; Li, Y.F.; Liu, C.; et al. Lipid metabolism dysfunction induced by age-dependent DNA methylation accelerates aging. Signal Transduct. Target. Ther. 2022, 7, 162. [Google Scholar] [CrossRef] [PubMed]
- Pahal, S.; Mainali, N.; Balasubramaniam, M.; Reis, R.J.S.; Ayyadevara, S. Mitochondria in aging and age associated diseases. Mitochondrion 2025, 82, 102022. [Google Scholar] [CrossRef]
- Sarparanta, J.; Garcia-Macia, M.; Singh, R. Autophagy and Mitochondria in Obesity and Type 2 Diabetes. Curr. Diabetes Rev. 2017, 13, 352–369. [Google Scholar] [CrossRef]
- Yang, J.S.; Lu, C.-C.; Kuo, S.-C.; Hsu, Y.M.; Tsai, S.C.; Chen, S.Y.; Chen, Y.T.; Lin, Y.J.; Huang, Y.C.; Chen, C.J.; et al. Autophagy and its link to type II diabetes mellitus. Biomedicine 2017, 7, 8. [Google Scholar] [CrossRef]
- Ou, Y.; Zhao, Y.-L.; Su, H. Pancreatic β-Cells, Diabetes and Autophagy. Endocr. Res. 2025, 50, 12–27. [Google Scholar] [CrossRef]
- Bays, H.E.; Toth, P.P.; Kris-Etherton, P.M.; Abate, N.; Aronne, L.J.; Brown, W.V.; Gonzalez-Campoy, J.M.; Jones, S.R.; Kumar, R.; La Forge, R.; et al. Obesity, adiposity, and dyslipidemia: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 304–383. [Google Scholar] [CrossRef]
- Grundy, S.M. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab. 2004, 89, 2595–2600. [Google Scholar] [CrossRef]
- Franssen, R.; Monajemi, H.; Stroes, E.S.; Kastelein, J.J. Obesity and dyslipidemia. Endocrinol. Metab. Clin. N. Am. 2011, 95, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common Neurodegenerative Pathways in Obesity, Diabetes, and Alzheimer’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hinder, L.M.; Callaghan, B.C.; Feldman, E.L. Neurological consequences of obesity. Lancet Neurol. 2017, 16, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Cai, D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol. Metab. 2013, 24, 40–47. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Neto, A.; Fernandes, A.; Barateiro, A. The complex relationship between obesity and neurodegenerative diseases: An updated review. Front. Cell Neurosci. 2023, 17, 1294420. [Google Scholar] [CrossRef]
- Hong, Y.; Yu, J.; Su, Y.; Mei, F.; Li, M.; Zhao, K.; Zhao, L.; Deng, W.H.; Chen, C.; Wang, W.X. High-fat diet aggravates acute pancreatitis via TLR4-mediated necroptosis and inflammation in rats. Oxid. Med. Cell Longev. 2020, 2020, 8172714. [Google Scholar] [CrossRef]
- Feng, Z.; Fang, C.; Ma, Y.; Chang, J. Obesity-induced blood-brain barrier dysfunction: Phenotypes and mechanisms. J. Neuroinflamm. 2024, 21, 110. [Google Scholar] [CrossRef]
- Hasel, P.; Aisenberg, W.H.; Bennett, F.C.; Liddelow, S.A. Molecular and metabolic heterogeneity of astrocytes and microglia. Cell Metab. 2023, 35, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Skarica, M.; Mansoor, M.; Bhandarkar, S.; Toro, S.; Pitt, D. Astrocyte heterogeneity in multiple sclerosis: Current understanding and technical challenges. Front. Cell Neurosci. 2021, 15, 726479. [Google Scholar] [CrossRef] [PubMed]
- Mazon, J.N.; de Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Pitt, D.; Lo, C.H.; Gauthier, S.A.; Hickman, R.A.; Longbrake, E.; Airas, L.M.; Mao-Draayer, Y.; Riley, C.; De Jager, P.L.; Wesley, S.; et al. Toward precision phenotyping of multiple sclerosis. Neurol—Neuroimmunol. Neuroinflamm. 2022, 9, e200025. [Google Scholar] [CrossRef]
- Bousquet, M.; St-Amour, I.; Vandal, M.; Julien, P.; Cicchetti, F.; Calon, F. High-fat diet exacerbates MPTP-induced dopaminergic degeneration in mice. Neurobiol. Dis. 2012, 45, 529–538. [Google Scholar] [CrossRef]
- Dugan, L.L.; Ali, S.S.; Shekhtman, G.; Roberts, A.J.; Lucero, J.; Quick, K.L.; Behrens, M.M. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 2009, 4, e5518. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Keller, J.N.; Morrison, C.D. Obesity vulnerability of the CNS. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2009, 1792, 395–400. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Longato, L.; Tong, M.; Wands, J.R. Insulin resistance and neurodegeneration: Roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr. Opin. Investig. Drugs 2009, 10, 1049–1060. [Google Scholar]
- An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Nieß, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. The role of gut-derived lipopolysaccharides and the intestinal barrier in fatty liver diseases. J. Gastrointest. Surg. 2022, 26, 671–683. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, Z.; Sun, G.; Zhang, D.; Zhang, J. Genetic evidence for the liver-brain axis: Lipid metabolism and neurodegenerative disease risk. Lipids Health Dis. 2025, 24, 41. [Google Scholar] [CrossRef]
- Davey Smith, G.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, Y.; Zhang, X.; Liu, L.; Zhou, B.; Mo, R.; Li, Y.; Li, H.; Li, F.; Tao, Y.; et al. Medium-chain triglycerides improved cognition and lipid metabolomics in mild to moderate Alzheimer’s disease patients with APOE4−/−: A double-blind, randomized, placebo-controlled crossover trial. Clin. Nutr. 2020, 39, 2092–2105. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sterling, N.W.; Du, G.; Sun, D.; Stetter, C.; Kong, L.; Zhu, Y.; Neighbors, J.; Lewis, M.M.; Chen, H.; et al. Brain cholesterol metabolism and Parkinson’s disease. Mov. Disord. 2019, 34, 386–395. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Sarno, A.; Romano, F.; Arianna, R.; Serpico, D.; Lavorgna, M.; Savastano, S.; Colao, A.; Di Somma, C. Lipid Metabolism and Statin Therapy in Neurodegenerative Diseases: An Endocrine View. Metabolites 2025, 15, 282. https://doi.org/10.3390/metabo15040282
Di Sarno A, Romano F, Arianna R, Serpico D, Lavorgna M, Savastano S, Colao A, Di Somma C. Lipid Metabolism and Statin Therapy in Neurodegenerative Diseases: An Endocrine View. Metabolites. 2025; 15(4):282. https://doi.org/10.3390/metabo15040282
Chicago/Turabian StyleDi Sarno, Antonella, Fiammetta Romano, Rossana Arianna, Domenico Serpico, Mariarosaria Lavorgna, Silvia Savastano, Annamaria Colao, and Carolina Di Somma. 2025. "Lipid Metabolism and Statin Therapy in Neurodegenerative Diseases: An Endocrine View" Metabolites 15, no. 4: 282. https://doi.org/10.3390/metabo15040282
APA StyleDi Sarno, A., Romano, F., Arianna, R., Serpico, D., Lavorgna, M., Savastano, S., Colao, A., & Di Somma, C. (2025). Lipid Metabolism and Statin Therapy in Neurodegenerative Diseases: An Endocrine View. Metabolites, 15(4), 282. https://doi.org/10.3390/metabo15040282