
From Childhood Obesity to Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) and Hyperlipidemia Through Oxidative Stress During Childhood

 ,
, 
Abstract
:1. Introduction
2. Obesity in Children—Scope of the Problem
2.1. Prevalence and Incidence
2.2. Etiology
2.3. Obesity, Metabolic Syndrome, and Chronic Systemic Inflammation
2.4. Hormones and Obesity
2.5. Associated Comorbidities of Childhood Obesity
3. Obesity and MASLD in Children—Clinical Diagnosis and Metabolic Consideration
3.1. Definition of Metabolic Dysfunction-Associated Steatotic Liver Disease MASLD
3.2. Genesis of MASLD
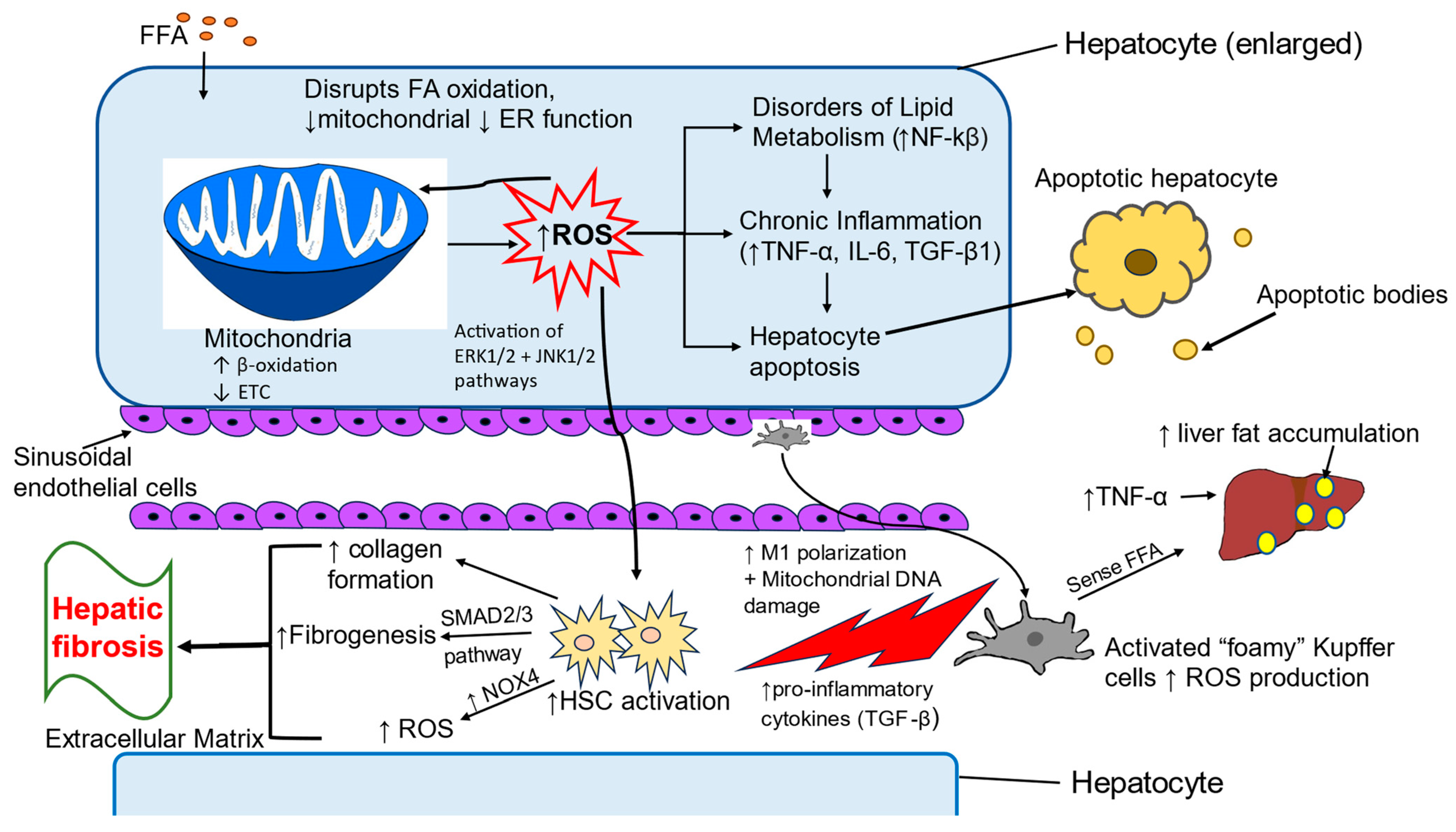
4. Oxidative Stress-Induced Pathologic Changes in MASLD
4.1. Oxidative Stress in Childhood MASLD
4.2. Liver Cells Involved in Oxidative Stress
4.2.1. Hepatocytes
4.2.2. Kupffer Cells
4.2.3. Hepatic Stellate Cells
4.3. Inflammatory Pathways Associated with Oxidative Stress
5. Treatments
5.1. Lifestyle Intervention Is the Primary Treatment for Pediatric MASLD
5.2. Medical Therapy
5.2.1. Metformin
5.2.2. GLP-1 Receptor Agonists
5.2.3. Resmetirom
5.2.4. Antioxidants
5.2.5. Vitamin D
5.2.6. Prebiotics and Probiotics
5.3. Surgical Intervention
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Serbis, A.; Giapros, V.; Galli-Tsinopoulo, A.; Siomou, E. Metabolic Syndrome in Children and Adolescents: Is There a Universally Accepted Definition? Does it Matter? Metab. Syndr. Relat. Disord. 2020, 18, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, P.; Weiskirchen, R. The Role of Obesity in Type 2 Diabetes Mellitus—An Overview. Int. J. Mol. Sci. 2024, 25, 1882. [Google Scholar] [CrossRef]
- Mainieri, F.; Tagi, V.M.; Chiarelli, F. Insulin resistance in children. Curr. Opin. Pediatr. 2022, 34, 400–406. [Google Scholar] [CrossRef]
- Lister, N.B.; Baur, L.A.; Felix, J.F.; Hill, A.J.; Marcus, C.; Reinehr, T.; Summerbell, C.; Wabitsch, M. Child and adolescent obesity. Nat. Rev. Dis. Primers 2023, 9, 24. [Google Scholar] [CrossRef]
- Newton, K.P.; Wilson, L.A.; Crimmins, N.A.; Fishbein, M.H.; Molleston, J.P.; Xanthakos, S.A.; Behling, C.; Schwimmer, J.B.; Nonalcoholic Steatohepatitis Clinical Research Network. Incidence of Type 2 Diabetes in Children With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, A.S.; Crowley, M.J.; Wang, Y.; Moylan, C.A.; Guy, C.D.; Henao, R.; Piercy, D.L.; Seymour, K.A.; Sudan, R.; Portenier, D.D.; et al. Glycemic control predicts severity of hepatocyte ballooning and hepatic fibrosis in nonalcoholic fatty liver disease. Hepatology 2021, 74, 1220–1233. [Google Scholar] [CrossRef]
- Alexopoulos, A.S.; Duffy, R.; Kobe, E.A.; German, J.; Moylan, C.A.; Soliman, D.; Jeffreys, A.S.; Coffman, C.J.; Crowley, M.J. Underrecognition of Nonalcoholic Fatty Liver Disease in Poorly Controlled Diabetes: A Call to Action in Diabetes Care. J. Endocr. Soc. 2021, 5, bvab155. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Ann. Hepatol. 2024, 29, 101133. [Google Scholar] [CrossRef]
- Vesa, C.M.; Behl, T.; Nemeth, S.; Bratu, O.G.; Diaconu, C.C.; Moleriu, R.D.; Negrut, N.; Zaha, D.C.; Bustea, C.; Radu, F.I.; et al. Prediction of NAFLD occurrence in prediabetes patients. Exp. Ther. Med. 2020, 20, 190. [Google Scholar] [CrossRef]
- Radu, F.; Potcovaru, C.-G.; Salmen, T.; Filip, P.V.; Pop, C.; Fierbințeanu-Braticievici, C. The Link between NAFLD and Metabolic Syndrome. Diagnostics 2023, 13, 614. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.Y.; Zhai, Z.Z.; Li, Z.F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem. Biol. Interact. 2020, 330, 109199. [Google Scholar] [CrossRef] [PubMed]
- Overi, D.; Carpino, G.; Franchitto, A.; Onori, P.; Gaudio, E. Hepatocyte injury and hepatic stem cell niche in the progression of non-alcoholic steatohepatitis. Cells 2020, 9, 590. [Google Scholar] [CrossRef]
- Sun, K.; Tordjman, J.; Clement, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Vily-Petit, J.; Soty-Roca, M.; Silva, M.; Raffin, M.; Gautier-Stein, A.; Rajas, F.; Mithieux, G. Intestinal gluconeogenesis prevents obesity-linked liver steatosis and non-alcoholic fatty liver disease. Gut 2020, 69, 2193–2202. [Google Scholar] [CrossRef]
- Soty, M.; Gautier-Stein, A.; Rajas, F.; Mithieux, G. Gut-Brain Glucose Signaling in Energy Homeostasis. Cell Metab. 2017, 25, 1231–1242. [Google Scholar] [CrossRef]
- Apperley, L.J.; Blackburn, J.; Erlandson-Parry, K.; Gait, L.; Laing, P.; Senniappan, S. Childhood obesity: A review of current and future management options. Clin. Endocrinol. 2022, 96, 288–301. [Google Scholar] [CrossRef]
- Jebeile, H.; Kelly, A.S.; O’Malley, G.; Baur, L.A. Obesity in children and adolescents: Epidemiology, causes, assessment, and management. Lancet Diabetes Endocrinol. 2022, 10, 351–365. [Google Scholar] [CrossRef]
- World Obesity Federation. Atlas of Childhood Obesity. Available online: https://s3-eu-west-1.amazonaws.com/wof-files/11996_Childhood_Obesity_Atlas_Report_ART_V2.pdf (accessed on 8 January 2024).
- Zhang, X.; Liu, J.; Ni, Y.; Yi, C.; Fang, Y.; Ning, Q.; Shen, B.; Zhang, K.; Liu, Y.; Yang, L.; et al. Global Prevalence of Overweight and Obesity in Children and Adolescents: A Systematic Review and Meta-Analysis. JAMA Pediatr. 2024, 178, 800–813. [Google Scholar] [CrossRef]
- Di Cesare, M.; Sorić, M.; Bovet, P.; Miranda, J.J.; Bhutta, Z.; Stevens, G.A.; Laxmaiah, A.; Kengne, A.P.; Bentham, J. The epidemiological burden of obesity in childhood: A worldwide epidemic requiring urgent action. BMC Med. 2019, 17, 212. [Google Scholar] [CrossRef]
- Arubuolawe, O.O.; Gabriel, O.T.; Anats, C.J.; Odion-Omonhimin, L.O.; Momodu, P.A.; Akanbi, S.A.; Babilsie, R.B.; Giri, K.; Okobi, O.E. Demographic and Socioeconomic Disparities in Adolescent Obesity: Insights From the National Survey of Children’s Health Database. Cureus 2024, 16, e72150. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Sanjeevi, R.R.; Balasubramanian, K.; Chahal, A.; Sharma, A.; Sidiq, M.A. Systematic Review on Prevalence of Overweight and Obesity among School Children and Adolescents in Indian Population. Indian J. Endocrinol. Metab. 2024, 28, 104–116. [Google Scholar] [CrossRef]
- Spinelli, A.; Buoncristiano, M.; Kovacs, V.A.; Yngve, A.; Spiroski, I.; Obreja, G.; Starc, G.; Pérez, N.; Rito, A.I.; Kunešová, M.; et al. Prevalence of Severe Obesity among Primary School Children in 21 European Countries. Obes. Facts. 2019, 12, 244–258. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef] [PubMed]
- Pop, T.L.; Maniu, D.; Rajka, D.; Lazea, C.; Cismaru, G.; Ştef, A.; Căinap, S.S. Prevalence of Underweight, Overweight and Obesity in School-Aged Children in the Urban Area of the Northwestern Part of Romania. Int. J. Environ. Res. Public Health 2021, 18, 5176. [Google Scholar] [CrossRef]
- Skinner, A.C.; Ravanbakht, S.N.; Skelton, J.A.; Perrin, E.M.; Armstrong, S.C. Prevalence of Obesity and Severe Obesity in US Children, 1999–2016. Pediatrics 2018, 141, e20173459. [Google Scholar] [CrossRef]
- Isong, I.A.; Rao, S.R.; Bind, M.A.; Avendaño, M.; Kawachi, I.; Richmond, T.K. Racial and Ethnic Disparities in Early Childhood Obesity. Pediatrics 2018, 141, e20170865. [Google Scholar] [CrossRef]
- Palit, S.; Sufyani, T.; Inungu, J.N.; Cheng, C.I.; Nartey, E. Behavioral Determinants of Childhood Obesity in the United States: An Exploratory Study. J. Obes. 2024, 2024, 9224425. [Google Scholar] [CrossRef]
- Dubois, L.; Ohm Kyvik, K.; Girard, M.; Fabiola Tatone-Tokuda, F.; Pérusse, D.; Hjelmborg, J.; Skytthe, A.; Rasmussen, F.; Wright, M.J.; Lichtenstein, P.; et al. Genetic and environmental contributions to weight, height, and BMI from birth to 19 years of age: An international study of over 12,000 twin pairs. PLoS ONE 2012, 2, e30153. [Google Scholar] [CrossRef]
- Carroll, J.E.; Emond, J.A.; VanKim, N.; Bertone-Johnson, E.; Sturgeon, S.R. A Latent Class Analysis of Family Eating Behaviors and Home Environment Habits on Preschool-Aged Children’s Body Mass Index. Child. Obes. 2024, 20, 643–652. [Google Scholar] [CrossRef]
- Chai, L.K.; Collins, C.; May, C.; Brain, K.; Wong See, D.; Burrows, T. Effectiveness of family-based weight management interventions for children with overweight and obesity: An umbrella review. JBI Database Syst. Rev. Implement. Rep. 2019, 17, 1341–1427. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Al-Ani, O.; Al-Ani, F. Children’s behaviour and childhood obesity. Zachowanie dzieci a otyłość dziecięca. Pediatr. Endocrinol. Diabetes Metab. 2024, 30, 148–158. [Google Scholar] [CrossRef]
- Kumar, S.; Kelly, A.S. Review of Childhood Obesity: From Epidemiology, Etiology, and Comorbidities to Clinical Assessment and Treatment. Mayo Clin. Proc. 2017, 92, 251–265. [Google Scholar] [CrossRef]
- Nogueira-de-Almeida, C.A.; Weffort, V.R.S.; Ued, F.D.V.; Ferraz, I.S.; Contini, A.A.; Martinez, E.Z.; Del Ciampo, L.A. What causes obesity in children and adolescents? J. Pediatr. 2024, 100 (Suppl. 1), S48–S56. [Google Scholar] [CrossRef] [PubMed]
- Bucher Della Torre, S.; Keller, A.; Laure Depeyre, J.; Kruseman, M. Sugar-Sweetened Beverages and Obesity Risk in Children and Adolescents: A Systematic Analysis on How Methodological Quality May Influence Conclusions. J. Acad. Nutr. Diet. 2016, 116, 638–659. [Google Scholar] [CrossRef]
- Monzani, A.; Ricotti, R.; Caputo, M.; Solito, A.; Archero, F.; Bellone, S.; Prodam, F. A Systematic Review of the Association of Skipping Breakfast with Weight and Cardiometabolic Risk Factors in Children and Adolescents. What Should We Better Investigate in the Future? Nutrients 2019, 11, 387. [Google Scholar] [CrossRef]
- Piątkowska-Chmiel, I.; Krawiec, P.; Ziętara, K.J.; Pawłowski, P.; Samardakiewicz, M.; Pac-Kożuchowska, E.; Herbetet, M. The Impact of Chronic Stress Related to COVID-19 on Eating Behaviors and the Risk of Obesity in Children and Adolescents. Nutrients 2023, 16, 54. [Google Scholar] [CrossRef]
- Costa, C.S.; Del-Ponte, B.; Assunção, M.C.F.; Santos, I.S. Consumption of ultra-processed foods and body fat during childhood and adolescence: A systematic review. Public Health Nutr. 2018, 21, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Wang, L.; Martín-Calvo, N.; Dhana, K.; Khandpur, N.; Rossato, S.L.; Steele, E.M.; Fung, T.T.; Chavarro, J.E.; Sun, Q.; et al. Ultraprocessed food intake and body mass index change among youths: A prospective cohort study. Am. J. Clin. Nutr. 2024, 120, 836–845. [Google Scholar] [CrossRef]
- Keller, A.; Bucher Della Torre, S. Sugar-Sweetened Beverages and Obesity among Children and Adolescents: A Review of Systematic Literature Reviews. Child. Obes. 2015, 11, 338–346. [Google Scholar] [CrossRef]
- Lara-Castor, L.; Micha, R.; Cudhea, F.; Miller, V.; Shi, P.; Zhang, J.; Sharib, J.R.; Erndt-Marino, J.; Cash, S.B.; Mozaffarian, D. Intake of sugar sweetened beverages among children and adolescents in 185 countries between 1990 and 2018: Population based study. BMJ 2024, 386, e079234. [Google Scholar] [CrossRef] [PubMed]
- Barnett, T.A.; Kelly, A.S.; Young, D.R.; Perry, C.K.; Pratt, C.A.; Edwards, N.M.; Rao, G.; Vos, M.B. Sedentary Behaviors in Today’s Youth: Approaches to the Prevention and Management of Childhood Obesity: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e142–e159. [Google Scholar] [CrossRef] [PubMed]
- Byun, D.; Kim, Y.; Jang, H.; Oh, H. Screen time and obesity prevalence in adolescents: An isotemporal substitution analysis. BMC Public Health 2024, 24, 3130. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.A.; Fiore, J.G.; Luo, M.; Kelly, S.; Adams, W.G.; Taveras, E.M.; Simione, M.; Kistin, C.J. Food and housing insecurity, COVID-19 pandemic effects on health-related activities, and care plans for children with obesity. Acad. Pediatr. 2024, 25, 102598. [Google Scholar] [CrossRef]
- Chang, T.H.; Chen, Y.C.; Chen, W.Y.; Chen, C.Y.; Hsu, W.Y.; Chou, Y.; Chang, Y.H. Weight gain associated with COVID-19 lockdown in children and adolescents: A systematic review and meta-analysis. Nutrients 2021, 13, 3668. [Google Scholar] [CrossRef]
- Jenssen, B.P.; Kelly, M.K.; Powell, M.; Bouchelle, Z.; Mayne, S.L.; Fiks, A.G. COVID-19 and Changes in Child Obesity. Pediatrics 2021, 147, e2021050123. [Google Scholar] [CrossRef]
- Ruiz-Roso, M.B.; de Carvalho Padilha, P.; Mantilla-Escalante, D.C.; Ulloa, N.; Brun, P.; Acevedo-Correa, D.; Arantes Ferreira Peres, W.; Martorell, M.; Aires, M.T.; de Oliveira Cardoso, L.; et al. COVID-19 Confinement and Changes of Adolescent’s Dietary Trends in Italy, Spain, Chile, Colombia and Brazil. Nutrients 2020, 12, 1807. [Google Scholar] [CrossRef]
- Pietrobelli, A.; Pecoraro, L.; Ferruzzi, A.; Heo, M.; Faith, M.; Zoller, T.; Antoniazzi, F.; Piacentini, G.; Fearnbach, S.N.; Heymsfield, S.B. Effects of COVID-19 Lockdown on Lifestyle Behaviors in Children with Obesity Living in Verona, Italy: A Longitudinal Study. Obesity 2020, 28, 1382–1385. [Google Scholar] [CrossRef]
- Wójcik, M.; Zachurzok, A. Obesity in children: Inheritance and treatment-state of art 2024. Pediatr. Endocrinol. Diabetes Metab. 2024, 30, 112–115. [Google Scholar] [CrossRef]
- Johnson, L.M. A genetic condition that spans both extremes of the nutritional spectrum. Pract. Lab. Med. 2024, 40, e00405. [Google Scholar] [CrossRef]
- Ahakoud, M.; Daha Belghiti, H.; Nedbour, A.; Bouramtane, A.; Chaouki, S.; Bouguenouch, L.; Ouldim, K. The Diagnosis and Genetic Mechanisms of Prader-Willi Syndrome: Findings From a Moroccan Population Study. Cureus 2023, 15, e37866. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, J.; Kinney, C.E.; Han, J.C. The Genetics of Obesity. Pediatr. Clin. N. Am. 2024, 71, 897–917. [Google Scholar] [CrossRef]
- Tillman, E.M.; Mertami, S. Precision medicine to identify, prevent, and treat pediatric obesity. Pharmacotherapy 2024, 44, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Waldrop, S.W.; Ibrahim, A.A.; Maya, J.; Monthe-Dreze, C.; Stanford, F.C. Overview of Pediatric Obesity as a Disease. Pediatr. Clin. N. Am. 2024, 71, 761–779. [Google Scholar] [CrossRef]
- Al-Hamad, D.; Raman, V. Metabolic syndrome in children and adolescents. Transl. Pediatr. 2017, 6, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Kachútová, I.; Hirošová, K.; Samohýl, M.; Mayer Vargová, K.; Babjaková, J.; Matejáková, L.; Argalášová, L.; Rimárová, K.; Dorko, E.; Jurkovičová, J. Continuous metabolic syndrome score in cardiovascular risk assessment in adolescents. Cent. Eur. J. Public Health 2025, 32, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Tamini , S.; Bondesan, A.; Caroli, D.; Marazzi, N.; Sartorio, A. The Ability of the Triglyceride-Glucose (TyG) Index and Modified TyG Indexes to Predict the Presence of Metabolic-Associated Fatty Liver Disease and Metabolic Syndrome in a Pediatric Population with Obesity. J. Clin. Med. 2025, 14, 2341. [Google Scholar] [CrossRef] [PubMed]
- Gepstein, V.; Weiss, R. Obesity as the Main Risk Factor for Metabolic Syndrome in Children. Front. Endocrinol. 2019, 10, 568. [Google Scholar] [CrossRef]
- Yang, K.; Song, M. New Insights into the Pathogenesis of Metabolic-Associated Fatty Liver Disease (MAFLD): Gut-Liver-Heart Crosstalk. Nutrients 2023, 15, 3970. [Google Scholar] [CrossRef]
- Kubota, N.; Kubota, T.; Kadowaki, T. Physiological and pathophysiological actions of insulin in the liver. Endocr. J. 2025, 72, 149–159. [Google Scholar] [CrossRef]
- Fujii, H.; Kawada, N.; Japan Study Group Of Nafld Jsg-Nafld. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Cusi, K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019, 1, 312–328. [Google Scholar] [CrossRef]
- Li, M.; Cui, M.; Li, G.; Liu, Y.; Xu, Y.; Eftekhar, S.P.; Ala, M. The Pathophysiological Associations Between Obesity, NAFLD, and Atherosclerotic Cardiovascular Diseases. Horm. Metab. Res. 2024, 56, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Szukiewicz, D. Molecular Mechanisms for the Vicious Cycle between Insulin Resistance and the Inflammatory Response in Obesity. Int. J. Mol. Sci. 2023, 24, 9818. [Google Scholar] [CrossRef]
- Engin, A. Reappraisal of Adipose Tissue Inflammation in Obesity. Adv. Exp. Med. Biol. 2024, 1460, 297–327. [Google Scholar]
- Calcaterra, V.; Regalbuto, C.; Porri, D.; Mazzon, E.; Vinci, F.; Zuccotti, G.; Fabiano, V.; Cena, H. Inflammation in Obesity-Related Complications in Children: The Protective Effect of Diet and Its Potential Role as a Therapeutic Agent. Biomolecules 2020, 10, 1324. [Google Scholar] [CrossRef]
- Kojta, I.; Chacińska, M.; Błachnio-Zabielska, A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients 2020, 12, 1305. [Google Scholar] [CrossRef]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier , J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.Y.; Han, Y.X.; Xu, S.N.; Jiang, H.L.; Wu, H.X.; Cai, J.M.; Li, L.; Bu, Y.H.; Xiao, F.; Liang, H.D.; et al. Adipose Tissue Plasticity: A Comprehensive Definition and Multidimensional Insight. Biomolecules 2024, 14, 1223. [Google Scholar] [CrossRef]
- Bence, K.K.; Birnbaum, M.J. Metabolic drivers of non-alcoholic fatty liver disease. Mol. Metab. 2021, 50, 101143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zheng, M.; Bai, R.; Chen, J.; Yang, H.; Luo, G. Molecular mechanisms of lipid droplets-mitochondria coupling in obesity and metabolic syndrome: Insights and pharmacological implications. Front. Physiol. 2024, 15, 1491815. [Google Scholar] [CrossRef]
- Shaunak, M.; Byrne, C.D.; Davis, N.; Afolabi, P.; Faust, S.N.; Davies, J.H.; Afolabi, P.; Faust, S.N.; Davies, J.H. Non-alcoholic fatty liver disease and childhood obesity. Arch. Dis. Child. 2021, 106, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; Lumeng, C.N. The initiation of metabolic inflammation in childhood obesity. J. Clin. Investig. 2017, 127, 65–73. [Google Scholar] [CrossRef]
- Yu, M.; Yu, B.; Chen, D. The effects of gut microbiota on appetite regulation and the underlying mechanisms. Gut Microbes 2024, 16, 2414796. [Google Scholar] [CrossRef]
- Howick, K.; Griffin, B.T.; Cryan, J.F.; Schellekens, H. From Belly to Brain: Targeting the Ghrelin Receptor in Appetite and Food Intake Regulation. Int. J. Mol. Sci. 2017, 18, 273. [Google Scholar] [CrossRef]
- Teixeira , M.R.; Silva, T.; Felício, , R.F.M.; Bozza, P.T.; Zembrzuski , V.M.; de Mello Neto, C.B.; da Fonseca, A.C.P.; Kohlrausch, F.B.; Salum, K.C.R. Exploring the genetic contribution in obesity: An overview of dopaminergic system genes. Behav. Brain Res. 2025, 480, 115401. [Google Scholar] [CrossRef] [PubMed]
- Lipid signatures of cardiometabolic risk in children and adolescents with obesity. Nat. Med. 2025, 31, 43–44. [CrossRef] [PubMed]
- Jing, L.; Nevius, C.D.; Friday, C.M.; Suever, J.D.; Pulenthiran, A.; Mejia-Spiegeler, A.; Kirchner, H.L.; Cochran, W.J.; Wehner, G.J.; Chishti, A.S.; et al. Ambulatory systolic blood pressure and obesity are independently associated with left ventricular hypertrophic remodeling in children. J. Cardiovasc. Magn. Reson. 2017, 19, 86. [Google Scholar] [CrossRef]
- Civilibal, T.N.; Oztarhan, K.; Bornaun, H.; Civilibal, A.M. Subclinical left ventricular structural and functional alterations in children with obesity: Is body mass or insulin resistance the main issue? Cardiol. Young 2024, 34, 2115–2121. [Google Scholar] [CrossRef]
- Crowley, D.I.; Khoury, P.R.; Urbina, E.M.; Ippisch, H.M.; Kimball, T.R. Cardiovascular impact of the pediatric obesity epidemic: Higher left ventricular mass is related to higher body mass index. J. Pediatr. 2011, 158, 709–714.e1. [Google Scholar] [CrossRef] [PubMed]
- Fazio, S.; Bellavite, P.; Affuso, F. Chronically Increased Levels of Circulating Insulin Secondary to Insulin Resistance: A Silent Killer. Biomedicines 2024, 12, 2416. [Google Scholar] [CrossRef]
- Pastore, I.; Bolla, A.M.; Montefusco, L.; Lunati, M.E.; Rossi, A.; Assi, E.; Zuccotti, G.V.; Fiorina, P. The Impact of Diabetes Mellitus on Cardiovascular Risk Onset in Children and Adolescents. Int. J. Mol. Sci. 2020, 21, 4928. [Google Scholar] [CrossRef]
- Bremer, A.A. Polycystic ovary syndrome in the pediatric population. Metab. Syndr. Relat. Disord. 2010, 8, 375–394. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhang, J.; Zhang, F.; Zhang, S.; Chen, X.; Liang, W.; Xie, Q. Resistance to the Insulin and Elevated Level of Androgen: A Major Cause of Polycystic Ovary Syndrome. Front. Endocrinol. 2021, 12, 741764. [Google Scholar] [CrossRef]
- AlBishi, L.; Alkhuraisi, L.S.; Alqahtani, M.M.; Alatawi, W.L.; Alghabban, A.T.; Anazi, M.H.; Aljohani, H.A.; Asseiri, R.A. Obstructive Sleep Apnea Among Obese Children in Tabuk City, Saudi Arabia. Cureus 2024, 16, e58714. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.L.; Zheng, Y.J.; Su, Z.; Wei, J.R.; Yang, Q.; Wang, C.C.; Li, J.H. Clinical features of obstructive sleep apnea in children with obesity. Chin. J. Contemp. Pediatr. 2021, 23, 933–937. [Google Scholar]
- Di Cicco, M.; Ghezzi, M.; Kantar, A.; Song, W.J.; Bush, A.; Peroni, D.; D’Auria, E. Pediatric obesity and severe asthma: Targeting pathways driving inflammation. Pharmacol. Res. 2023, 188, 106658. [Google Scholar] [CrossRef]
- Rinonapoli, G.; Pace, V.; Ruggiero, C.; Ceccarini, P.; Bisaccia, M.; Meccariello, L.; Caraffa, A. Obesity and bone: A complex relationship. Int. J. Mol. Sci. 2021, 22, 13662. [Google Scholar] [CrossRef]
- Lotan, R.; Thein, R.; Gordon, B.; Tenenbaum, S.; Derazne, E.; Tzur, D.; Afek, A.; Hershkovich, O. Is There an Association between BMI, Height, and Gender and Long-Bone Fractures during Childhood and Adolescence? A Large Cross-Sectional Population Study of 911,206 Subjects. Children 2023, 10, 984. [Google Scholar] [CrossRef]
- Perry, D.C.; Metcalfe, D.; Lane, S.; Turner, S. Childhood Obesity and Slipped Capital Femoral Epiphysis. Pediatrics 2018, 142, e20181067. [Google Scholar] [CrossRef]
- Farella, I.; Chiarito, M.; Vitale, R.; D’Amato, G.; Faienza, M.F. The “Burden” of Childhood Obesity on Bone Health: A Look at Prevention and Treatment. Nutrients 2025, 17, 491. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Kalkwarf, H.J.; Hornung, L.; Siegel, R.; Arce-Clachar, A.C.; Sheridan, R.; Ippisch, H.M.; Xanthakos, S.A. Histologic Severity of Nonalcoholic Fatty Liver Disease Associates with Reduced Bone Mineral Density in Children. Dig. Dis. Sci. 2023, 68, 644–655. [Google Scholar] [CrossRef]
- Avinun, R.; Hariri, A.R. A polygenic score for body mass index is associated with depressive symptoms via early life stress: Evidence for gene-environment correlation. J. Psychiatr.Res. 2019, 118, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.C.; Lam, J.M.; Barankin, B.; Leong, K.F.; Hon, K.L. Acanthosis Nigricans: An Updated Review. Curr. Pediatr. Rev. 2022, 19, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Çağ, Y.; Sağer, S.G.; Akçay, M.; Kaytan, İ.; Söbü, E.; Erdem, A.; Akın, Y. The relationship between body mass index and cerebrospinal fluid pressure in children with pseudotumor cerebri. Ital. J. Pediatr. 2024, 50, 150. [Google Scholar] [CrossRef]
- Yu, E.L.; Golshan, S.; Harlow, K.E.; Angeles, J.E.; Durelle, J.; Goyal, N.P.; Newton, K.P.; Sawh, M.C.; Hooker, J.; Sy, E.Z.; et al. Prevalence of Nonalcoholic Fatty Liver Disease in Children with Obesity. J. Pediatr. 2019, 207, 64–70. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J.; Valenti, L.; Davidson, N.O. Genetic Pathways in Nonalcoholic Fatty Liver Disease: Insights From Systems Biology. Hepatology 2020, 72, 330–346. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Lemieux, I.; Després, J.P. Metabolic Syndrome: Past, Present and Future. Nutrients 2020, 12, 3501. [Google Scholar] [CrossRef] [PubMed]
- Stroes, A.R.; Vos, M.; Benninga, M.A.; Koot, B.G.P. Pediatric MASLD: Current understanding and practical approach. Eur. J. Pediatr. 2024, 184, 29. [Google Scholar] [CrossRef]
- Loomba, R.; Wong, V.W. Implications of the new nomenclature of steatotic liver disease and defi-nition of metabolic dysfunction-associated steatotic liver disease. Aliment. Pharmacol. Ther. 2024, 59, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Abrams, S.H.; Barlow, S.E.; Caprio, S.; Daniels, S.R.; Kohli, R.; Mouzaki, M.; Sathya, P.; Schwimmer, J.B.; Sundaram, S.S.; et al. NASPGHAN clinical practice guideline for the diagnosis and treatment of nonalcoholic fatty liver disease in children: Recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroen-terology, Hepatology and Nutrition (NASPGHAN). J. Pediatr. Gastroenterol. Nutr. 2017, 64, 319–334. [Google Scholar]
- Canivet, C.M.; Faure, S. Diagnostic et évaluation de l’hépatopathie stéatosique métabolique. Rev. Méd. Interne 2024, 45, 41–47. [Google Scholar] [CrossRef]
- Fernández, J.R.; Redden, D.T.; Pietrobelli, A.; Allison, D.B. Waist circumference percentiles in nationally representative samples of African-American, European-American, and Mexican-American children and adolescents. J. Pediatr. 2004, 145, 439–444. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, M.H.; Mogren, C.; Gleason, T.; Stevens, W.R. Relationship of Hepatic Steatosis to Adipose Tissue Distribution in Pediatric Nonalcoholic Fatty Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2006, 42, 83–88. [Google Scholar] [CrossRef]
- Agbaje, A.O. Waist-circumference-to-height-ratio had better longitudinal agreement with DEXA-measured fat mass than BMI in 7237 children. Pediatr. Res. 2024, 96, 1369–1380. [Google Scholar] [CrossRef]
- Zhang, L.; El-Shabrawi, M.; Baur, L.A.; Byrne, C.D.; Targher, G.; Kehar, M.; Porta, G.; Lee, W.S.; Lefere, S.; Turan, S.; et al. An international multidisciplinary consensus on pediatric metabolic dysfunction-associated fatty liver disease. Med 2024, 5, 797–815.e2. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alisi, A.; Valenti, L.; Miele, L.; Feldstein, A.E.; Alkhouri, N. NAFLD in children: New genes, new diagnostic modalities and new drugs. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Birnbaum, M.J. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. 2021, 33, 2329–2354. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Dunn, W.; Norman, G.J.; Pardee, P.E.; Middleton, M.S.; Kerkar, N.; Sirlin, C.B. SAFETY study: Alanine aminotransferase cutoff values are set too high for reliable detection of pediatric chronic liver disease. Gastroenterology 2010, 138, 1357–1364. [Google Scholar] [CrossRef]
- Spengler, E.K.; Loomba, R. Recommendations for Diagnosis, Referral for Liver Biopsy, and Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Mayo Clin. Proc. 2015, 90, 1233–1246. [Google Scholar] [CrossRef]
- Anderson, E.L.; Howe, L.D.; Fraser, A.; Callaway, M.P.; Sattar, N.; Day, C.; Tilling, K.; Lawlor, D.A. Weight trajectories through infancy and childhood and risk of non-alcoholic fatty liver disease in adolescence: The ALSPAC study. J. Hepatol. 2014, 61, 626–632. [Google Scholar] [CrossRef]
- Northstone, K.; Lewcock, M.; Groom, A.; Boyd, A.; Macleod, J.; Timpson, N.; Wells, N. The Avon Longitudinal Study of Parents and Children (ALSPAC): An update on the enrolled sample of index children in 2019. Wellcome Open Res. 2019, 4, 51. [Google Scholar] [CrossRef]
- Barrera, F.; George, J. The role of diet and nutritional intervention in managing patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef]
- Papadopoulos, G.; Legaki, A.I.; Georgila, K.; Vorkas, P.; Giannousi, E.; Stamatakis, G.; Moustakas, I.I.; Petrocheilou, M.; Pyrina, I.; Gercken, B.; et al. Integrated omics analysis for characterization of the contribution of high fructose corn syrup to non-alcoholic fatty liver disease in obesity. Metabolism 2023, 144, 155552. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Tovoli, F.; Negrini, G.; Farì, R.; Guidetti, E.; Faggiano, C.; Napoli, L.; Bolondi, L.; Granito, A. Increased risk of nonalcoholic fatty liver disease in patients with coeliac disease on a gluten-free diet: Beyond traditional metabolic factors. Aliment. Pharmacol. Ther. 2018, 48, 538–546. [Google Scholar] [CrossRef]
- Wang, Z.; Tan, W.; Huang, J.; Li, Q.; Wang, J.; Su, H.; Guo, C.; Liu, H. Small intestinal bacterial overgrowth and metabolic dysfunction-associated steatotic liver disease. Front. Nutr. 2024, 11, 1502151. [Google Scholar] [CrossRef]
- Valvano, M.; Longo, S.; Stefanelli, G.; Frieri, G.; Viscido, A.; Latella, G. Celiac Disease, Gluten-Free Diet, and Metabolic and Liver Disorders. Nutrients 2020, 12, 940. [Google Scholar] [CrossRef]
- Goyal, N.P.; Rosenthal, S.B.; Nasamran, C.; Behling, C.A.; Angeles, J.E.; Fishbein, M.H.; Harlow, K.E.; Jain, A.K.; Molleston, J.P.; Newton, K.P.; et al. Nonalcoholic fatty liver disease risk and histologic severity are associated with genetic polymorphisms in children. Hepatology 2022, 77, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants. Biomolecules 2020, 10, 1702. [Google Scholar] [CrossRef] [PubMed]
- Zarkovic, N. Roles and Functions of ROS and RNS in Cellular Physiology and Pathology. Cells 2020, 9, 767. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Pirgon, Ö.; Bilgin, H.; Çekmez, F.; Kurku, H.; Dündar, B.N. Association between insulin resistance and oxidative stress parameters in obese adolescents with non-alcoholic fatty liver disease. J. Clin. Res. Pediatr. Endocrinol. 2013, 5, 33–39. [Google Scholar]
- Jakubek, P.; Kalinowski, P.; Karkucinska-Wieckowska, A.; Kaikini, A.M.; Simões, I.C.; Potes, Y.; Kruk, B.; Grajkowska, W.; Pinton, P.; Milkiewicz, P.; et al. Oxidative stress in metabolic dysfunction-associated steatotic liver disease (MASLD): How does the animal model resemble human disease? FASEB J. 2024, 38, e23466. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, A.; Duseja, A.; Das, A.; Dhiman, R.K.; Chawla, Y.K.; Kohli, K.K.; Bhansali, A. Patients with Nonalcoholic Fatty Liver Disease (NAFLD) have Higher Oxidative Stress in Comparison to Chronic Viral Hepatitis. J. Clin. Exp. Hepatol. 2013, 3, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.Q.D.; Rocha, R.; Daltro, C.H.D.C.; Andrade, S.C.S.; Cotrim, H.P. Serum glutathione peroxidase is associated with nonalcoholic fatty liver disease in children and adolescents. La glutatión-peroxidasa sérica se asocia a la enfermedad del hígado graso no alcohólico en niños y adolescentes. Nutr. Hosp. 2024, 41, 1165–1171. [Google Scholar]
- Mandala, A.; Janssen, R.C.; Palle, S.; Short, K.R.; Friedman, J.E. Pediatric Non-Alcoholic Fatty Liver Disease: Nutritional Origins and Potential Molecular Mechanisms. Nutrients 2020, 12, 3166. [Google Scholar] [CrossRef] [PubMed]
- Kopiczko, N.; Bobrus-Chociej, A.; Harasim-Symbor, E.; Tarasów, E.; Wojtkowska, M.; Chabowski, A.; Lebensztejn, D.M. Serum concentration of fatty acids in children with obesity and nonalcoholic fatty liver disease. Nutrition 2022, 94, 111541. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Y.; Xu, C.; Hong, Y.; Lu, H.; Wu, J.; Chen, Y. Association between serum free fatty acid levels and nonalcoholic fatty liver disease: A cross-sectional study. Sci. Rep. 2014, 4, 5832. [Google Scholar] [CrossRef]
- Yang, M.; Gong, S.; Ye, S.Q.; Lyman, B.; Geng, L.; Chen, P.; Li, D.-Y. Non-Alcoholic Fatty Liver Disease in Children: Focus on Nutritional Interventions. Nutrients 2014, 6, 4691–4705. [Google Scholar] [CrossRef]
- Nobili, V.; Alisi, A.; Mosca, A.; Crudele, A.; Zaffina, S.; Denaro, M.; Smeriglio, A.; Trombetta, D. The Antioxidant Effects of Hydroxytyrosol and Vitamin E on Pediatric Nonalcoholic Fatty Liver Disease, in a Clinical Trial: A New Treatment? Antioxid. Redox Signal. 2019, 31, 127–133. [Google Scholar] [CrossRef]
- Negri, R.; Trinchese, G.; Carbone, F.; Caprio, M.G.; Stanzione, G.; di Scala, C.; Micillo, T.; Perna, F.; Tarotto, L.; Gelzo, M.; et al. Randomised Clinical Trial: Calorie Restriction Regimen with Tomato Juice Supplementation Ameliorates Oxidative Stress and Preserves a Proper Immune Surveillance Modulating Mitochondrial Bioenergetics of T-Lymphocytes in Obese Children Affected by Non-Alcoholic Fatty Liver Disease (NAFLD). J. Clin. Med. 2020, 9, 141. [Google Scholar] [CrossRef] [PubMed]
- Spahis, S.; Alvarez, F.; Ahmed, N.; Dubois, J.; Jalbout, R.; Paganelli, M.; Grzywacz, K.; Delvin, E.; Peretti, N.; Levy, E. Non-alcoholic fatty liver disease severity and metabolic complications in obese children: Impact of omega-3 fatty acids. J. Nutr. Biochem. 2018, 58, 28–36. [Google Scholar] [CrossRef]
- Gong, J.; Tu, W.; Liu, J.; Tian, D. Hepatocytes: A key role in liver inflammation. Front. Immunol. 2023, 13, 1083780. [Google Scholar] [CrossRef]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Baeck, C.; Tacke, F. Balance of inflammatory pathways and interplay of immune cells in the liver during homeostasis and injury. EXCLI J. 2014, 13, 67–81. [Google Scholar] [PubMed]
- Allameh, A.; Niayesh-Mehr, R.; Aliarab, A.; Sebastiani, G.; Pantopoulos, K. Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants 2023, 12, 1653. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2142. [Google Scholar] [CrossRef]
- de Mochel, N.S.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef]
- Niranjan, S.; Phillips, B.E.; Giannoukakis, N. Uncoupling hepatic insulin resistance-hepatic inflammation to improve insulin sensitivity and to prevent impaired metabolism-associated fatty liver disease in type 2 diabetes. Front. Endocrinol. 2023, 14, 1193373. [Google Scholar] [CrossRef]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, G.; Heo, S.-Y.; Roh, Y.-S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2022, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Yin, J.; Huang, K. Free Fatty Acids Increase Intracellular Lipid Accumulation and Oxidative Stress by Modulating PPARα and SREBP-1c in L-02 Cells. Lipids 2016, 51, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef]
- Tang, S.; Geng, Y.; Lin, Q. The role of mitophagy in metabolic diseases and its exercise intervention. Front. Physiol. 2024, 15, 1339128. [Google Scholar] [CrossRef]
- Gandhi, C.R. Oxidative Stress and Hepatic Stellate Cells: A PARADOXICAL RELATIONSHIP. Trends Cell. Mol. Biol. 2012, 7, 1–10. [Google Scholar]
- Wang, K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 2014, 16, e996. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef]
- Xu, H.L.; Wan, S.R.; An, Y.; Wu, Q.; Xing, Y.H.; Deng, C.H.; Zhang, P.P.; Long, Y.; Xu, B.T.; Jiang, Z.Z. Targeting cell death in NAFLD: Mechanisms and targeted therapies. Cell Death Discov. 2024, 10, 399. [Google Scholar] [CrossRef]
- Park, S.J.; Garcia Diaz, J.; Um, E.; Hahn, Y.S. Major roles of kupffer cells and macrophages in NAFLD development. Front. Endocrinol. 2023, 14, 1150118. [Google Scholar] [CrossRef] [PubMed]
- Slevin, E.; Baiocchi, L.; Wu, N.; Ekser, B.; Sato, K.; Lin, E.; Ceci, L.; Chen, L.; Lorenzo, S.R.; Xu, W.; et al. Kupffer Cells: Inflammation Pathways and Cell-Cell Interactions in Alcohol-Associated Liver Disease. Am. J. Pathol. 2020, 190, 2185–2193. [Google Scholar] [CrossRef]
- Handa, P.; Vemulakonda, A.; Kowdley, K.V.; Uribe, M.; Méndez-Sánchez, N. Mitochondrial DNA from hepatocytes as a ligand for TLR9: Drivers of nonalcoholic steatohepatitis? World J. Gastroenterol. 2016, 22, 6965–6971. [Google Scholar] [CrossRef] [PubMed]
- Diehl, K.L.; Vorac, J.; Hofmann, K.; Meiser, P.; Unterweger, I.; Kuerschner, L.; Weighardt, H.; Förster, I.; Thiele, C. Kupffer Cells Sense Free Fatty Acids and Regulate Hepatic Lipid Metabolism in High-Fat Diet and Inflammation. Cells 2020, 9, 2258. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Novo, E.; Busletta, C.; Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef]
- Zhou, Y.; Long, D.; Zhao, Y.; Li, S.; Liang, Y.; Wan, L.; Zhang, J.; Xue, F.; Feng, L. Oxidative stress-mediated mitochondrial fission promotes hepatic stellate cell activation via stimulating oxidative phosphorylation. Cell Death Dis. 2022, 13, 689. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving Concepts in the Pathogenesis of NASH: Beyond Steatosis and Inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Xu, X.; Poulsen, K.L.; Wu, L.; Liu, S.; Miyata, T.; Song, Q.; Wei, Q.; Zhao, C.; Lin, C.; Yang, J. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct. Target. Ther. 2022, 7, 287. [Google Scholar] [CrossRef]
- Guilliams, M.; Scott, C.L. Liver macrophages in health and disease. Immunity 2022, 55, 1515–1529. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Srinivas, A.N.; Suresh, D.; Santhekadur, P.K.; Suvarna, D.; Kumar, D.P. Extracellular Vesicles as Inflammatory Drivers in NAFLD. Front. Immunol. 2021, 11, 627424. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; He, S.; Mao, X.; Zhang, Y.; Cai, Y.; Li, S. Effect of Hepatic Macrophage Polarization and Apoptosis on Liver Ischemia and Reperfusion Injury During Liver Transplantation. Front. Immunol. 2020, 11, 1193. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Choi, H.; Kim, H.J.; Lee, M.O. Chemotactic cytokines secreted from Kupffer cells contribute to the sex-dependent susceptibility to non-alcoholic fatty liver diseases in mice. Life Sci. 2022, 306, 120846. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, X.J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e20. [Google Scholar] [CrossRef]
- Kaufmann, B.; Kui, L.; Reca, A.; Leszczynska, A.; Kim, A.D.; Booshehri, L.M.; Wree, A.; Friess, H.; Hartmann, D.; Broderick, L.; et al. Cell-specific Deletion of NLRP3 Inflammasome Identifies Myeloid Cells as Key Drivers of Liver Inflammation and Fibrosis in Murine Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2024, 14, 751–767. [Google Scholar] [CrossRef]
- Ramos-Tovar, E.; Muriel, P. NLRP3 inflammasome in hepatic diseases: A pharmacological target. Biochem. Pharmacol. 2023, 217, 115861. [Google Scholar] [CrossRef]
- Osman, H.A.; Abuhamdah, S.M.A.; Hassan, M.H.; Hashim, A.A.; Ahmed, A.E.; Elsayed, S.S.; El-Sawy, S.A.; Gaber, M.A.; Abdelhady, M. NLRP3 inflammasome pathway involved in the pathogenesis of metabolic associated fatty liver disease. Sci. Rep. 2024, 14, 19648. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.R. TLR9 in MAFLD and NASH: At the Intersection of Inflammation and Metabolism. Front. Endocrinol. 2021, 11, 613639. [Google Scholar] [CrossRef] [PubMed]
- Smirne, C.; Croce, E.; Di Benedetto, D.; Cantaluppi, V.; Comi, C.; Sainaghi, P.P.; Minisini, R.; Grossini, E.; Pirisi, M. Oxidative Stress in Non-Alcoholic Fatty Liver Disease. Livers 2022, 2, 30–76. [Google Scholar] [CrossRef]
- de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. Int. J. Mol. Sci. 2020, 21, 3858. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.P.; Mao, X.L.; Chen, Y.H.; Yan, L.L.; Ye, L.P.; Li, S.W. Reactive Oxygen Species Induce Fatty Liver and Ischemia-Reperfusion Injury by Promoting Inflammation and Cell Death. Front. Immunol. 2022, 13, 870239. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Lee, S.M.; Koh, D.H.; Jun, D.W.; Roh, Y.J.; Kang, H.T.; Oh, J.H.; Kim, H.S. Auranofin attenuates hepatic steatosis and fibrosis in nonalcoholic fatty liver disease via NRF2 and NF- κB signaling pathways. Clin. Mol. Hepatol. 2022, 28, 827–840. [Google Scholar] [CrossRef]
- Jiang, J.X.; Török, N.J. NADPH Oxidases in Chronic Liver Diseases. Adv. Hepatol. 2014, 2014, 742931. [Google Scholar] [CrossRef]
- Nascè, A.; Gariani, K.; Jornayvaz, F.R.; Szanto, I. NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook. Antioxidants 2022, 11, 1131. [Google Scholar] [CrossRef]
- McGlinchey, A.J.; Govaere, O.; Geng, D.; Ratzieu, V.; Allison, M.; Bousier, J.; Petta, S.; de Oliviera, C.; Bugianesi, E.; Schattenberg, J.M.; et al. Metabolic signatures across the full spectrum of non-alcoholic fatty liver disease. JHEP Rep. 2022, 4, 100477. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Herranz-Itúrbide, M.; Peñuelas-Haro, I.; Espinosa-Sotelo, R.; Bertran, E.; Fabregat, I. The TGF-β/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells 2021, 10, 2312. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef]
- Afarin, R.; Behdarvand, T.; Shakerian, E.; Salehipour Bavarsad, S.; Rashidi, M. Exosomes of Whartons’ jelly mesenchymal stem cell reduce the NOX genes in TGF-β-induced hepatic fibrosis. Iran. J. Basic. Med. Sci. 2022, 25, 1498–1503. [Google Scholar] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Asadizade, S.; Hatami, M.; Salehipour Bavarsad, S.; Kabizade, B.; Shakerian, E.; Rashidi, M. Curcumin Modulates NOX Gene Expression and ROS Production via P-Smad3C in TGF-β-Activated Hepatic Stellate Cells. Iran. Biomed. J. 2024, 28, 31–37. [Google Scholar] [CrossRef]
- Wilhelmsen, I.; Combriat, T.; Dalmao-Fernandez, A.; Stokowiec, J.; Wang, C.; Olsen, P.A.; Wik, J.A.; Boichuk, Y.; Aizenshtadt, A.; Krauss, S. The effects of TGF-β-induced activation and starvation of vitamin A and palmitic acid on human stem cell-derived hepatic stellate cells. Stem Cell Res. Ther. 2024, 15, 223. [Google Scholar] [CrossRef]
- Garvey, W.T.; Mechanick, J.I.; Brett, E.M.; Garber, A.J.; Hurley, D.L.; Jastreboff, A.M.; Nadolsky, K.; Pessah-Pollack, R.; Plodkowski, R.; Reviewers of the AACE/ACE Obesity Clinical Practice Guidelines. American Association of Clinical Endocrinologists and American College of Endocrinology comprehensive clinical practice guidelines for medical care of patients with obesity. Endocr. Pract. 2016, 22, 1–203. [Google Scholar]
- Marchesini, G.; Petta, S.; Dalle Grave, R. Diet, weight loss, and liver health in nonalcoholic fatty liver disease: Pathophysiology, evidence, and practice. Hepatology 2016, 63, 2032–2043. [Google Scholar] [CrossRef]
- Deshmukh, A.; Sood, V.; Lal, B.B.; Khanna, R.; Alam, S.; Sarin, S.K. Effect of Indo-Mediterranean diet versus calorie-restricted diet in children with non-alcoholic fatty liver disease: A pilot randomized control trial. Pediatr. Obes. 2024, 19, e13163. [Google Scholar] [CrossRef]
- Flores Lopez, A.G.; Quiros-Tejeira, R.E.; Lyden, E.; McGill, B.; Dike, C.R. Association between BMI Change, Transaminases, and Other Metabolic Parameters in Children with Nonalcoholic Fatty Liver Disease. J. Obes. 2024, 2024, 6997280. [Google Scholar] [CrossRef] [PubMed]
- Serbis, A.; Polyzos, S.A.; Paschou, S.A.; Siomou, E.; Kiortsis, D.N. Diet, exercise, and supplements: What is their role in the management of the metabolic dysfunction-associated steatotic liver disease in children? Endocrine 2024, 85, 988–1006. [Google Scholar] [CrossRef]
- Cuda, S.E.; Kharofa, R.; Williams, D.R.; O’Hara, V.; Conroy, R.; Karjoo, S.; Paisley, J.; Censani, M.; Browne, N.T. Metabolic, behavioral health, and disordered eating comorbidities associated with obesity in pediatric patients: An Obesity Medical Association (OMA) Clinical Practice Statement 2022. Obes. Pillars. 2022, 3, 100031. [Google Scholar] [CrossRef]
- Lefere, S.; Dupont, E.; De Guchtenaere, A.; Van Biervliet, S.; Vande Velde, S.; Verhelst, X.; Devisscher, L.; Van Vlierberghe, H.; Geerts, A.; De Bruyne, R. Intensive Lifestyle Management Improves Steatosis and Fibrosis in Pediatric Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2022, 20, 2317–2326.e4. [Google Scholar] [CrossRef] [PubMed]
- Xanthakos, S.A.; Lavine, J.E.; Yates, K.P.; Schwimmer, J.B.; Molleston, J.P.; Rosenthal, P.; Murray, K.F.; Vos, M.B.; Jain, A.K.; Scheimann, A.O.; et al. Progression of fatty liver disease in children receiving standard of care lifestyle advice. Gastroenterology 2020, 159, 1731–1751.e10. [Google Scholar] [CrossRef]
- González-Ruiz, K.; Ramírez-Vélez, R.; Correa-Bautista, J.E.; Peterson, M.D.; García-Hermoso, A. The Effects of Exercise on Abdominal Fat and Liver Enzymes in Pediatric Obesity: A Systematic Review and Meta-Analysis. Child. Obes. 2017, 13, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, V.; Magenes, V.C.; Vandoni, M.; Berardo, C.; Marin, L.; Bianchi, A.; Cordaro, E.; Silvestro, G.S.; Silvestri, D.; Carnevale Pellino, V.; et al. Benefits of Physical Exercise as Approach to Prevention and Reversion of Non-Alcoholic Fatty Liver Disease in Children and Adolescents with Obesity. Children 2022, 9, 1174. [Google Scholar] [CrossRef]
- Sood, V.; Alam, S.; Nagral, A.; Srivastava, A.; Deshmukh, A.; Bavdekar, A.; Acharyya, B.C.; Geetha, S.M.; Gupte, G.; Bhatia, I.; et al. Practice Recommendations for Metabolic Dysfunction-Associated Steatotic Liver Disease by the Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition (ISPGHAN). Indian Pediatr. 2024, 61, 919–934. [Google Scholar]
- Medrano, M.; Cadenas-Sanchez, C.; Álvarez-Bueno, C.; Cavero-Redondo, I.; Ruiz, J.R.; Ortega, F.B.; Labayen, I. Evidence-Based Exercise Recommendations to Reduce Hepatic Fat Content in Youth- a Systematic Review and Meta-Analysis. Prog. Cardiovasc. Dis. 2018, 61, 222–231. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Piester, T.L.; Jagtap, N.; Kalapala, R. Review of paediatric obesity and non-alcoholic fatty liver disease-A focus on emerging non-pharmacologic treatment strategies. Pediatr. Obes. 2023, 18, e13067. [Google Scholar] [CrossRef] [PubMed]
- Koutoukidis, D.A.; Koshiaris, C.; Henry, J.A.; Noreik, M.; Morris, E.; Manoharan, I.; Tudor, K.; Bodenham, E.; Dunnigan, A.; Jebb, S.A.; et al. The effect of the magnitude of weight loss on non-alcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2021, 115, 15445. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, E.A.; Evans, C.V.; Burda, B.U.; Walsh, E.S.; Eder, M.; Lozano, P. Screening for obesity and intervention for weight management in children and adolescents: Evidence report and systematic review for the US Preventive Services Task Force. JAMA 2017, 317, 2427–2444. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, T.; Toschke, A.M. Onset of puberty and cardiovascular risk factors in untreated obese children and adolescents: A 1-year follow-up study. Arch. Pediatr. Adolesc. Med. 2009, 163, 709–715. [Google Scholar] [CrossRef]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef]
- Yasuda, N.; Inoue, T.; Nagakura, T.; Yamazaki, K.; Kira, K.; Saeki, T.; Tanaka, I. Metformin causes reduction of food intake and body weight gain and improvement of glucose intolerance in combination with dipeptidyl peptidase IV inhibitor in Zucker fa/fa rats. J. Pharmacol. Exp. Ther. 2004, 310, 614–619. [Google Scholar] [CrossRef]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef]
- Abrosimov, R.; Baeken, M.W.; Hauf, S.; Wittig, I.; Hajieva, P.; Perrone, C.E.; Moosmann, B. Mitochondrial complex I inhibition triggers NAD+-independent glucose oxidation via successive NADPH formation, "futile" fatty acid cycling, and FADH2 oxidation. Geroscience 2024, 46, 3635–3658. [Google Scholar] [CrossRef]
- Pinyopornpanish, K.; Leerapun, A.; Pinyopornpanish, K.; Chattipakorn, N. Effects of metformin on hepatic steatosis in adults with nonalcoholic fatty liver disease and diabetes: Insights from the cellular to patient levels. Gut Liver. 2021, 15, 827–840. [Google Scholar] [CrossRef]
- Maslak, E.; Zabielski, P.; Kochan, K.; Kus, K.; Jasztal, A.; Sitek, B.; Proniewski, B.; Wojcik, T.; Gula, K.; Kij, A.; et al. The liver-selective NO donor, V-PYRRO/NO, protects against liver steatosis and improves postprandial glucose tolerance in mice fed high fat diet. Biochem. Pharmacol. 2015, 93, 389–400. [Google Scholar] [CrossRef]
- Anggreini, P.; Kuncoro, H.; Sumiwi, S.A.; Levita, J. Role of the AMPK/SIRT1 pathway in non-alcoholic fatty liver disease (Review). Mol. Med. Rep. 2023, 27, 35. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, T.; Rahman, M.M.; Khan, F.; Kabir, F.; Nahar, K.; Lasker, S.; Islam, M.D.; Hossain, M.M.; Hasan, R.; Rana, S.; et al. Metformin treatment reverses high fat diet- induced non-alcoholic fatty liver diseases and dyslipidemia by stimulating multiple and anti-inflammatory pathways. Biochem. Biophys. Rep. 2021, 28, 101168. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.; Hernández-Arriaga, A.; Kehm, R.; Sánchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, X.; Cong, B. Advances in the mechanism of metformin with wide-ranging effects on regulation of the intestinal microbiota. Front. Microbiol. 2024, 15, 1396031. [Google Scholar] [CrossRef]
- Theodoridis, X.; Kalopitas, G.; Vadarlis, A.; Bakaloudi, D.R.; Gkiourtzis, Ν.; Dionysopoulos, G.; Karanika, E.; Tsekitsidi, E.; Chourdakis, M. Comparative efficacy of different treatment modalities in the management of pediatric non-alcoholic fatty liver disease: A systematic review and network meta-analysis. Pharm. Ther. 2022, 240, 108294. [Google Scholar] [CrossRef]
- Akcam, M.; Boyaci, A.; Pirgon, O.; Kaya, S.; Uysal, S.; Dundar, B.N. Therapeutic effect of metformin and vitamin E versus prescriptive diet in obese adolescents with fatty liver. Int. J. Vitam. Nutr. Res. 2011, 81, 398–406. [Google Scholar] [CrossRef]
- Nobili, V.; Manco, M.; Ciampalini, P.; Alisi, A.; Devito, R.; Bugianesi, E.; Marcellini, M.; Marchesini, G. Metformin use in children with nonalcoholic fatty liver disease: An open-label, 24-month, observational pilot study. Clin. Ther. 2008, 30, 1168–1176. [Google Scholar] [CrossRef]
- Mann, J.P.; Tang, G.Y.; Nobili, V.; Armstrong, M.J. Evaluations of lifestyle, dietary, and pharmacologic treatments for pediatric nonalcoholic fatty liver disease: A systematic review. Clin. Gastroenterol. Hepatol. 2019, 17, 1457–1476.e7. [Google Scholar] [CrossRef]
- Gkiourtzis, N.; Michou, P.; Moutafi, M.; Glava, A.; Cheirakis, K.; Christakopoulos, A.; Vouksinou, E.; Fotoulaki, M. The benefit of metformin in the treatment of pediatric non-alcoholic fatty liver disease: A systematic review and meta-analysis of randomized controlled trials. Eur. J. Pediatr. 2023, 182, 4795–4806. [Google Scholar] [CrossRef]
- Said, A.; Akhter, A. Meta-analysis of randomized controlled trials of pharmacologic agents in non-alcoholic steatohepatitis. Ann. Hepatol. 2017, 16, 538–547. [Google Scholar] [CrossRef]
- Alfaris, N.; Waldrop, S.; Johnson, V.; Boaventura, B.; Kendrick, K.; Stanford, F.C. GLP-1 single, dual, and triple receptor agonists for treating type 2 diabetes and obesity: A narrative review. EClinicalMedicine 2024, 75, 102782. [Google Scholar] [CrossRef] [PubMed]
- Byberg, S.; Holt, J.; Sandsdal, R.M.; Holm, L.A.; Madsen, L.B.; Christensen, B.J.; Jensen, S.B.K.; Hansen, T.; Holm, J.C.; Torekov, S. Protocol for a randomised, double-blinded, controlled trial of youth with childhood-onset obesity treated with semaglutide 2.4 mg/week: The RESETTLE trial. BMJ 2024, 14, e082446. [Google Scholar] [CrossRef] [PubMed]
- Shettar, V.; Patel, S.; Kidambi, S. Epidemiology of obesity and pharmacologic treatment options. Nutr. Clin. Pract. 2017, 32, 441–462. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes-state-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef]
- Stefan, N.; Haring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Patel Chavez, C.; Cusi, K.; Kadiyala, S. The emerging role of glucagon-like peptide-1 receptor agonists for the management of NAFLD. J. Clin. Endocrinol. Metab. 2022, 107, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Gong, Y.; Jiang, M.; Gao, Y.; Guo, S.; Huo, J.; Zhao, Z.; Li, C. Utilization of glucagon-like peptide-1 receptor agonists in children and adolescents in China: A real-world study. Front. Endocrinol. 2023, 14, 1170127. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef]
- Blundell, J.; Finlayson, G.; Axelsen, M.; Flint, A.; Gibbons, C.; Kvist, T.; Hjerpsted, J.B. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes. Metab. 2017, 19, 1242–1251. [Google Scholar] [CrossRef]
- Qin, W.; Yang, J.; Deng, C.; Ruan, Q.; Duan, K. Efficacy and safety of semaglutide 2.4 mg for weight loss in overweight or obese adults without diabetes: An updated systematic review and meta-analysis including the 2-year STEP 5 trial. Diabetes Obes. Metab. 2024, 26, 911–923. [Google Scholar] [CrossRef]
- Christou, G.A.; Katsiki, N.; Blundell, J.; Fruhbeck, G.; Kiortsis, D.N. Semaglutide as a promising antiobesity drug. Obes. Rev. 2019, 20, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.S.; Rudser, K.D.; Nathan, B.M.; Fox, C.K.; Metzig, A.M.; Coombes, B.J.; Fitch, A.K.; Bomberg, E.M.; Abuzzahab, M.J. The effect of glucagon-like peptide-1 receptor agonist therapy on body mass index in adolescents with severe obesity: A randomized, placebo-controlled, clinical trial. JAMA Pediatr. 2013, 167, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Ramirez Tovar, A.; He, Z.; Soler Rodriguez, D.M.; Vos, M.B.; Arora, S.; Fadoju, D. Glucagon-like Peptide-1 Receptor Agonists-A Potential New Medication for Pediatric Metabolic-Dysfunction-Associated Steatotic Liver Disease (MASLD). Children 2024, 11, 275. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.; Grohs, M.; El Gammal, A.T.; Wolter, S.; Lehnert, H.; Mann, O.; Mittag, J.; Kirchner, H. Reduced expression of thyroid hormone receptor β in human nonalcoholic steatohepatitis. Endocr. Connect. 2018, 7, 1448–1456. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef]
- Harrison, S.A.; Taub, R.; Neff, G.W.; Lucas, K.J.; Labriola, D.; Moussa, S.E.; Alkhouri, N.; Bashir, M.R. Resmetirom for nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled phase 3 trial. Nat. Med. 2023, 29, 2919–2928. [Google Scholar] [CrossRef]
- Raza, S.; Tewari, A.; Rajak, S.; Sinha, R.A. Vitamins and non-alcoholic fatty liver disease: A Molecular Insight. Liver Res. 2021, 5, 62–71. [Google Scholar] [CrossRef]
- Dong, J.X.; Jiang, L.L.; Liu, Y.P.; Zheng, A.X. Association between composite dietary antioxidant index and metabolic dysfunction-associated fatty liver disease: A cross-sectional study from NHANES. BMC Gastroenterol. 2024, 24, 465. [Google Scholar] [CrossRef]
- Nagashimada, M.; Ota, T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB Life 2019, 71, 516–522. [Google Scholar] [CrossRef]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of vitamin E for nonalcoholic steatohepatitis in patients with type 2 diabetes: A randomized controlled trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Abera, M.; Suresh, S.B.; Malireddi, A.; Boddeti, S.; Noor, K.; Ansar, M.; Malasevskaia, I. Vitamin E and Non-alcoholic Fatty Liver Disease: Investigating the Evidence Through a Systematic Review. Cureus 2024, 16, e72596. [Google Scholar] [CrossRef]
- Ando, Y.; Jou, J.H. Nonalcoholic fatty liver disease and recent guideline updates. Clin. Liver Dis. 2021, 17, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhang, Q.; Wang, J.; Li, Y.; Wang, X.; Bao, Y. Vitamin E synthesis and response in plants. Front. Plant Sci. 2022, 13, 994058. [Google Scholar] [CrossRef]
- El Hadi, H.; Vettor, R.; Rossato, M. Vitamin E as a treatment for nonalcoholic fatty liver disease: Reality or myth? Antioxidants 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Panera, N.; Braghini, M.R.; Crudele, A.; Smeriglio, A.; Bianchi, M.; Condorelli, A.G.; Nobili, R.; Conti, L.A.; De Stefanis, C.; Lioci, G.; et al. Combination Treatment with Hydroxytyrosol and Vitamin E Improves NAFLD-Related Fibrosis. Nutrients 2022, 14, 3791. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, D.; Hamid, Z.T. The role of vitamin D in diabetes and cardiovascular disease: An updated review of the literature. Dis. Markers 2015, 2015, 580474. [Google Scholar] [CrossRef]
- Guo, X.F.; Wang, C.; Yang, T.; Li, S.; Li, K.L.; Li, D. Vitamin D and non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Food Funct. 2020, 11, 7389–7399. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Wang, J.; Chen, X.; Reichetzeder, C.; Elitok, S.; Krämer, B.K.; Doebis, C.; Huesker, K.; von Baehr, V.; et al. Vitamin D Is Associated with Lipid Metabolism: A Sex- and Age-Dependent Analysis of a Large Outpatient Cohort. Nutrients 2024, 16, 3936. [Google Scholar] [CrossRef]
- Latic, N.; Erben, R.G. Vitamin D and cardiovascular disease, with emphasis on hypertension, atherosclerosis, and heart failure. Int. J. Mol. Sci. 2020, 21, 6483. [Google Scholar] [CrossRef]
- Szymczak-Pajor, I.; Śliwińska, A. Analysis of association between vitamin D deficiency and insulin resistance. Nutrients 2019, 11, 794. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Jiang, Z.; Song, H.; Zhang, Y.; Chen, H.; Liu, W.; Wei, X.; Li, L.; Li, W.; Li, X. Vitamin D supplementation alleviates high fat diet-induced metabolic associated fatty liver disease by inhibiting ferroptosis pathway. Eur. J. Nutr. 2024, 64, 50. [Google Scholar] [CrossRef] [PubMed]
- Knuth, M.M.; Xue, J.; Elnagheeb, M.; Gharaibeh, R.Z.; Schoenrock, S.A.; McRitchie, S.; Brouwer, C.; Sumner, S.J.; Tarantino, L.; Valdar, W.; et al. Early life exposure to vitamin D deficiency impairs molecular mechanisms that regulate liver cholesterol biosynthesis, energy metabolism, inflammation, and detoxification. Front. Endocrinol. 2024, 15, 1335855. [Google Scholar] [CrossRef] [PubMed]
- Hariri, M.; Zohdi, S. Effect of vitamin D on non-alcoholic fatty liver disease: A systematic review of randomized controlled clinical trials. Int. J. Prev. Med. 2019, 10, 14. [Google Scholar] [CrossRef]
- Park, C.Y.; Shin, S.; Han, S.N. Multifaceted Roles of Vitamin D for Diabetes: From Immunomodulatory Functions to Metabolic Regulations. Nutrients 2024, 16, 3185. [Google Scholar] [CrossRef]
- Maestro, B.; Molero, S.; Bajo, S.; Dávila, N.; Calle, C. Transcriptional activation of the human insulin receptor gene by 1,25-dihydroxyvitamin D3. Cell Biochem. Funct. 2002, 20, 227–232. [Google Scholar] [CrossRef]
- Mirhosseini, N.; Vatanparast, H.; Mazidi, M.; Kimball, S.M. Vitamin D Supplementation, Glycemic Control, and Insulin Resistance in Prediabetics: A Meta-Analysis. J. Endocr. Soc. 2018, 2, 687–709. [Google Scholar] [CrossRef]
- Sindhughosa, D.A.; Wibawa, I.D.N.; Mariadi, I.K.; Somayana, G. Additional treatment of vitamin D for improvement of insulin resistance in non-alcoholic fatty liver disease patients: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 7716. [Google Scholar] [CrossRef]
- Pramono, A.; Jocken, J.; Blaak, E.E.; van Baak, M.A. The effect of vitamin D supplementation on insulin sensitivity: A systematic review and meta-analysis. Diabetes Care 2020, 43, 1659–1669. [Google Scholar] [CrossRef]
- El Amrousy, D.; Abdelhai, D.; Shawky, D. Vitamin D and nonalcoholic fatty liver disease in children: A randomized controlled clinical trial. Eur. J. Pediatr. 2022, 181, 579–586. [Google Scholar] [CrossRef]
- Makri, E.; Goulas, A.; Polyzos, S.A. Epidemiology, Pathogenesis, Diagnosis and Emerging Treatment of Nonalcoholic Fatty Liver Disease. Arch. Med. Res. 2021, 52, 25–37. [Google Scholar] [CrossRef] [PubMed]
- de Faria Ghetti, F.; Oliveira, D.G.; de Oliveira, J.M.; de Castro Ferreira, L.E.V.V.; Cesar, D.E.; Moreira, A.P.B. Influence of gut microbiota on the development and progression of nonalcoholic steatohepatitis. Eur. J. Nutr. 2018, 57, 861–876. [Google Scholar] [CrossRef]
- Chopyk, D.M.; Grakoui, A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology 2020, 159, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Loman, B.R.; Hernández-Saavedra, D.; An, R.; Rector, R.S. Prebiotic and probiotic treatment of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Nutr. Rev. 2018, 76, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Borka Balas, R.; Meliț, L.E.; Lupu, A.; Lupu, V.V.; Mărginean, C.O. Prebiotics, Probiotics, and Synbiotics-A Research Hotspot for Pediatric Obesity. Microorganisms 2023, 11, 2651. [Google Scholar] [CrossRef]
- Castillo, V.; Figueroa, F.; González-Pizarro, K.; Jopia, P.; Ibacache-Quiroga, C. Probiotics and Prebiotics as a Strategy for Non-Alcoholic Fatty Liver Disease, a Narrative Review. Foods 2021, 10, 1719. [Google Scholar] [CrossRef]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Radovanovic Spurnic, A.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N.; et al. Gut-Liver Axis, Gut Microbiota, and Its Modulation in the Management of Liver Diseases: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef]
- Yarahmadi, A.; Afkhami, H.; Javadi, A.; Kashfi, M. Understanding the complex function of gut microbiota: Its impact on the pathogenesis of obesity and beyond: A comprehensive review. Diabetol. Metab. Syndr. 2024, 16, 308. [Google Scholar] [CrossRef]
- Beisner, J.; Filipe Rosa, L.; Kaden-Volynets, V.; Stolzer, I.; Günther, C.; Bischoff, S.C. Prebiotic Inulin and Sodium Butyrate Attenuate Obesity-Induced Intestinal Barrier Dysfunction by Induction of Antimicrobial Peptides. Front. Immunol. 2021, 12, 678360. [Google Scholar] [CrossRef]
- Imai, N.; Cohen, D.E. Trimming the Fat: Acetyl-CoA Carboxylase Inhibition for the Management of NAFLD. Hepatology 2018, 68, 2062–2065. [Google Scholar] [CrossRef]
- Hu, H.; Lin, A.; Kong, M.; Yao, X.; Yin, M.; Xia, H.; Ma, J.; Liu, H. Intestinal microbiome and NAFLD: Molecular insights and therapeutic perspectives. J. Gastroenterol. 2020, 55, 142–158. [Google Scholar] [CrossRef]
- Eisenberg, D.; Shikora, S.A.; Aarts, E.; Aminian, A.; Angrisani, L.; Cohen, R.V.; de Luca, M.; Faria, S.L.; Goodpaster, K.P.S.; Haddad, A.; et al. 2022 American Society of Metabolic and Bariatric Surgery (ASMBS) and International Federation for the Surgery of Obesity and Metabolic Disorders (IFSO) Indications for Metabolic and Bariatric Surgery. Obes. Surg. 2023, 33, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Defining Childhood Weight Status BMI for Children and Teens. Available online: https://www.cdc.gov/bmi/child-teen-calculator/bmi-categories.html?CDC_AAref_Val=https://www.cdc.gov/obesity/basics/childhood-defining.html (accessed on 17 December 2021).
- Armstrong, S.; Lazorick, S.; Hampl, S.; Skelton, J.A.; Wood, C.; Collier, D.; Perrin, E.M. Physical examination findings among children and adolescents with obesity: An evidence-based review. Pediatrics 2016, 137, e20151766. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Mosca, A.; De Peppo, F.; Caccamo, R.; Cutrera, R.; Giordano, U.; De Stefanis, C.; Alisi, A.; Baumann, U.; Silecchia, G.; et al. The benefit of sleeve gastrectomy in obese adolescents on nonalcoholic steatohepatitis and hepatic fibrosis. J. Pediatr. 2017, 180, 31–37.e2. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Mosca, A.; De Peppo, F.; Caccamo, R.; Cutrera, R.; Giordano, U.; De Stefanis, C.; Alisi, A.; Baumann, U.; Silecchia, G.; et al. Indications and Limitations of Bariatric Intervention in Severely Obese Children and Adolescents With and Without Nonalcoholic Steatohepatitis: ESPGHAN Hepatology Committee Position Statement. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 550–561. [Google Scholar]
- Brichta, C.; Fishbein, M.; Ryder, J.R. Outcomes of adolescent bariatric surgery: Liver disease. Surg. Obes. Relat. Dis. 2025, 21, 9–15. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cause | Examples | References |
|---|---|---|
| Exogenous | Excessive caloric intake, sedentary lifestyle, insufficient sleep, increased screen time, parenting styles, lack of school resources, community factors, ethnicity | [18,20,31,32,33,34] |
| Monogenic | Leptin deficiency, leptin receptor gene deficiency, POMC deficiency, MC4R gene mutation, PCSK1 deficiency, SIM 1 deficiency, KSR2 deficiency, NTRK2 mutations, BDNF mutations | [53] |
| Syndromes | Albright hereditary osteodystrophy, Prader–Willi syndrome, Down syndrome, Turner syndrome, Bardet–Biedl syndrome, WAGR syndrome, Fragile X syndrome, Cohen syndrome, Beckwith–Wiedemann syndrome | [50,51,52,53] |
| Endocrine | Hypothyroidism, Cushing syndrome, growth hormone deficiency, hyperinsulinism, PCOS | [18,35] |
| Medications | Glucocorticoids, antipsychotics, beta blockers, lithium, antidepressants, antiepileptics, diabetes medications | [18,35] |
| Neurologic | Traumatic brain injury, neoplasms, post-cranial radiation, hypothalamic obesity | [35,54,55] |
| Psychiatric | Depression, eating disorders | [18,20,35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Accacha, S.; Barillas-Cerritos, J.; Srivastava, A.; Ross, F.; Drewes, W.; Gulkarov, S.; De Leon, J.; Reiss, A.B. From Childhood Obesity to Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) and Hyperlipidemia Through Oxidative Stress During Childhood. Metabolites 2025, 15, 287. https://doi.org/10.3390/metabo15050287
Accacha S, Barillas-Cerritos J, Srivastava A, Ross F, Drewes W, Gulkarov S, De Leon J, Reiss AB. From Childhood Obesity to Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) and Hyperlipidemia Through Oxidative Stress During Childhood. Metabolites. 2025; 15(5):287. https://doi.org/10.3390/metabo15050287
Chicago/Turabian StyleAccacha, Siham, Julia Barillas-Cerritos, Ankita Srivastava, Frances Ross, Wendy Drewes, Shelly Gulkarov, Joshua De Leon, and Allison B. Reiss. 2025. "From Childhood Obesity to Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) and Hyperlipidemia Through Oxidative Stress During Childhood" Metabolites 15, no. 5: 287. https://doi.org/10.3390/metabo15050287
APA StyleAccacha, S., Barillas-Cerritos, J., Srivastava, A., Ross, F., Drewes, W., Gulkarov, S., De Leon, J., & Reiss, A. B. (2025). From Childhood Obesity to Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) and Hyperlipidemia Through Oxidative Stress During Childhood. Metabolites, 15(5), 287. https://doi.org/10.3390/metabo15050287