
Integrated Metabolomics and Lipidomics Analysis Reveals the Mechanism Behind the Action of Chiglitazar on the Protection Against Sepsis-Induced Acute Lung Injury

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs and Materials
2.2. Animal Model
2.3. Samplings
2.4. Tissue Weighing and Histochemical Analysis
2.5. Immunohistochemical Analysis
2.6. Cell Culture
2.7. Cell Viability Assay
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Metabolomics Methodology
2.10. Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
3.1. Chiglitazar Improved the Survival Rate of CLP Rats and Reduced Inflammatory Cytokines in BALF
3.2. Chiglitazar Ameliorated Acute Lung Injury in Septic Rats
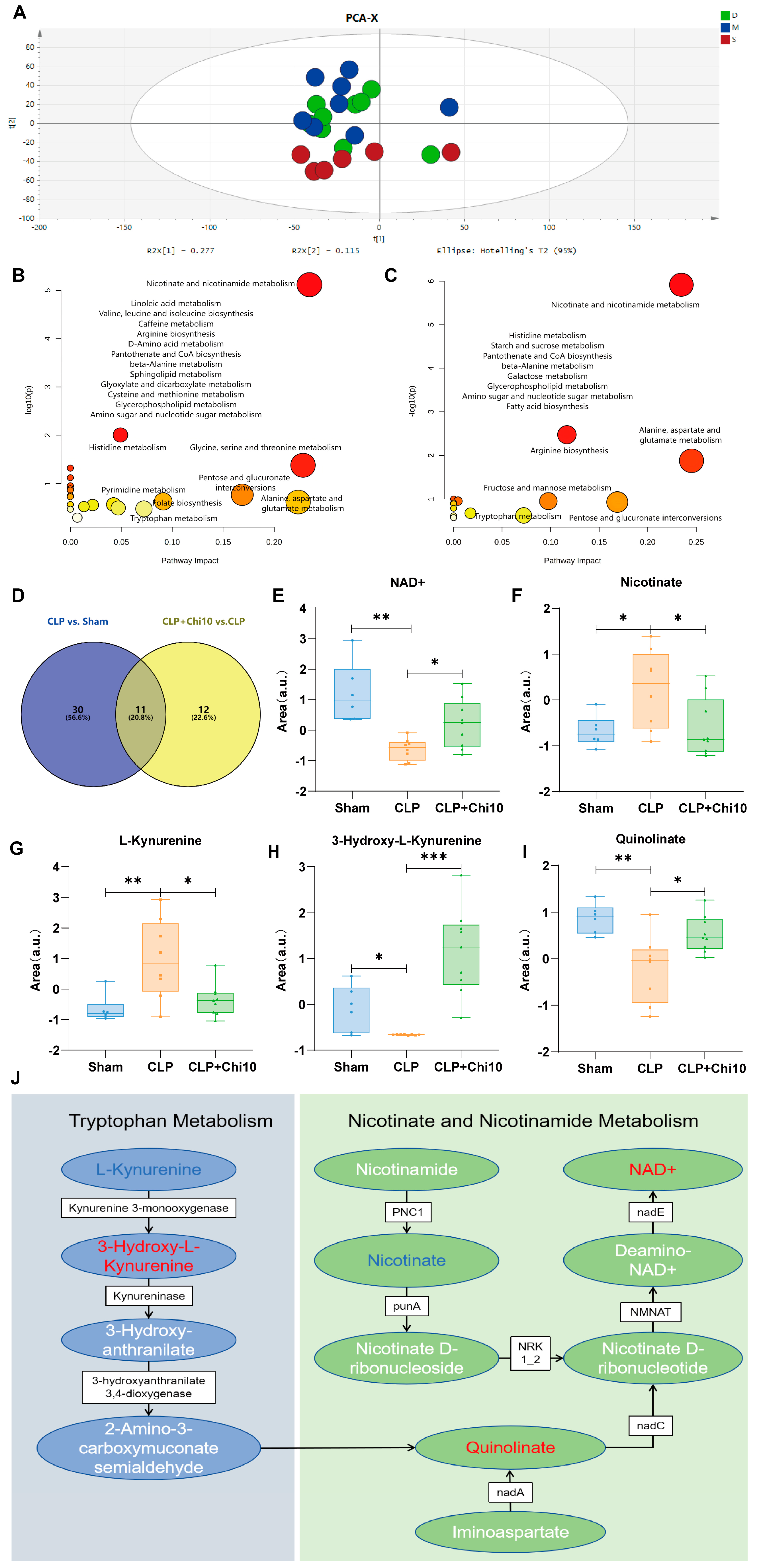
3.3. Chiglitazar Induced the Reprogramming of Kynurenine–Nicotinamide Pathway in Septic Rats
3.4. Considerable Dysregulations of Lipid Metabolism Found in Septic Rats Were Reversed by the Pretreatment of Chiglitazar
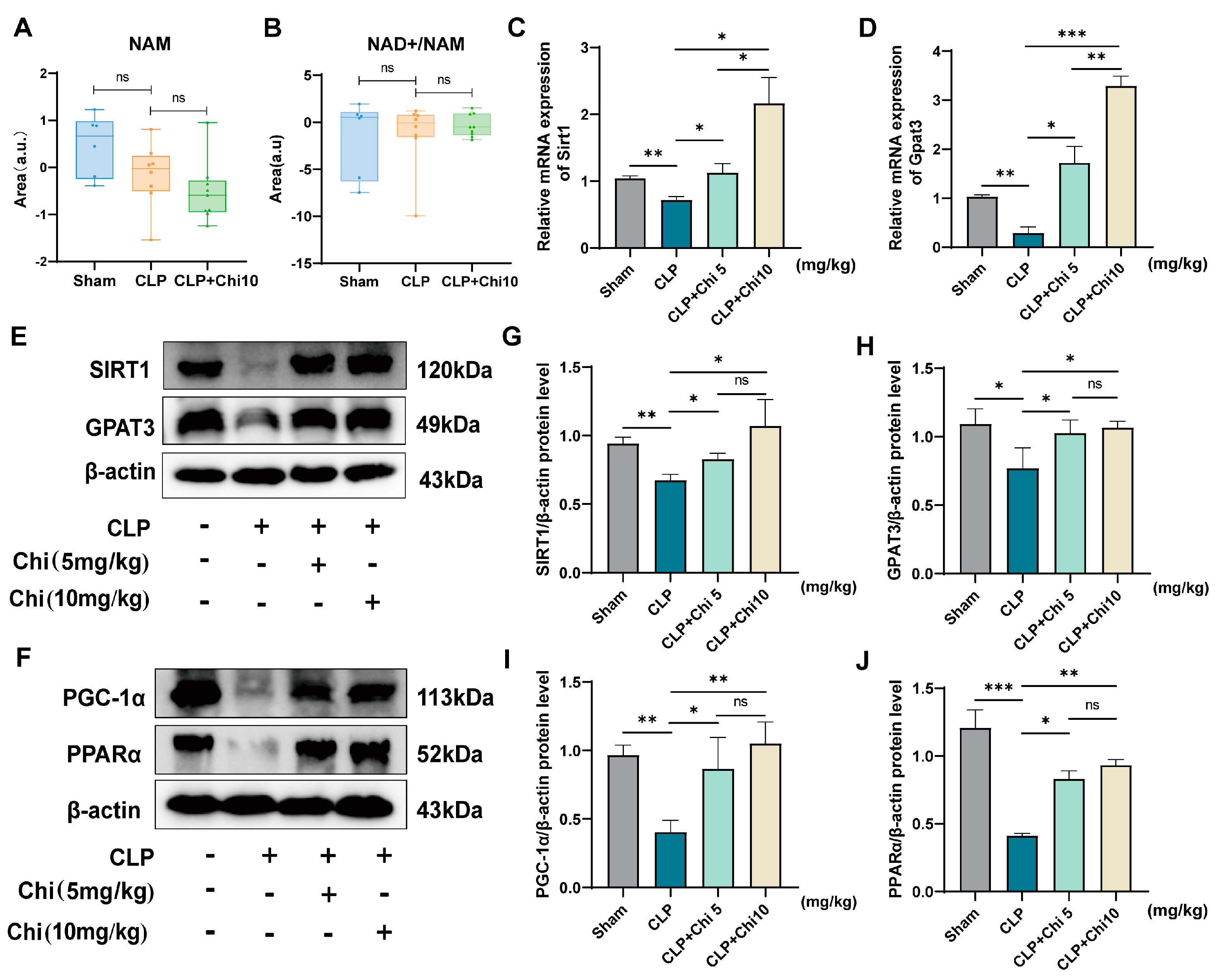
3.5. Chiglitazar Pretreatment Ameliorates Sepsis-Induced Metabolic Dysregulation via Activation of the SIRT1/PGC-1α/PPARα/GPAT3 Signaling Axis
3.6. SIRT1 Inhibitor EX-527 Reversed the Activation of Signaling Pathways by Chiglitazar in BEAS-2B Cells
3.7. The SIRT1 Inhibitor EX-527 Reversed the Effect of Chiglitazar on Sepsis-Induced Lung Injury
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Hao, C.; Tang, S. From sepsis to acute respiratory distress syndrome (ARDS): Emerging preventive strategies based on molecular and genetic researches. Biosci. Rep. 2020, 40, BSR20200830. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Lei, M.; Zhang, J.; Kang, H.; Liu, H.; Zhou, F. Acute lung injury caused by sepsis: How does it happen? Front. Med. 2023, 10, 1289194. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, N.; Routledge, M.; Schlebusch, S.; Lipman, J.; Conway Morris, A. Antimicrobial-associated harm in critical care: A narrative review. Intensive Care Med. 2020, 46, 225–235. [Google Scholar] [CrossRef]
- Annane, D.; Renault, A.; Brun-Buisson, C.; Megarbane, B.; Quenot, J.P.; Siami, S.; Cariou, A.; Forceville, X.; Schwebel, C.; Martin, C.; et al. Hydrocortisone plus Fludrocortisone for Adults with Septic Shock. N. Engl. J. Med. 2018, 378, 809–818. [Google Scholar] [CrossRef]
- Liu, D.; Huang, S.Y.; Sun, J.H.; Zhang, H.C.; Cai, Q.L.; Gao, C.; Li, L.; Cao, J.; Xu, F.; Zhou, Y.; et al. Sepsis-induced immunosuppression: Mechanisms, diagnosis and current treatment options. Mil. Med. Res. 2022, 9, 56. [Google Scholar] [CrossRef]
- Wasyluk, W.; Zwolak, A. Metabolic Alterations in Sepsis. J. Clin. Med. 2021, 10, 2412. [Google Scholar] [CrossRef]
- Liu, W.; Liu, T.; Zheng, Y.; Xia, Z. Metabolic Reprogramming and Its Regulatory Mechanism in Sepsis-Mediated Inflammation. J. Inflamm. Res. 2023, 16, 1195–1207. [Google Scholar] [CrossRef]
- Fitzpatrick, S.F. Immunometabolism and Sepsis: A Role for HIF? Front. Mol. Biosci. 2019, 6, 85. [Google Scholar] [CrossRef]
- Addissouky, T.A.; El Tantawy El Sayed, I.; Ali, M.M.A.; Wang, Y.; El Baz, A.; Khalil, A.A.; Elarabany, N. Molecular Pathways in Sepsis Pathogenesis: Recent Advances and Therape utic Avenues. J. Cell. Immunol. 2023, 5, 174–183. [Google Scholar] [CrossRef]
- Lu, X.; Li, G.; Liu, Y.; Luo, G.; Ding, S.; Zhang, T.; Li, N.; Geng, Q. The role of fatty acid metabolism in acute lung injury: A special focus on immunometabolism. Cell. Mol. Life Sci. CMLS 2024, 81, 120. [Google Scholar] [CrossRef]
- Muniz-Santos, R.; Lucieri-Costa, G.; de Almeida, M.A.P.; Moraes-de-Souza, I.; Brito, M.; Silva, A.R.; Gonçalves-de-Albuquerque, C.F. Lipid oxidation dysregulation: An emerging player in the pathophysiology of sepsis. Front. Immunol. 2023, 14, 1224335. [Google Scholar] [CrossRef]
- Barker, G.; Leeuwenburgh, C.; Brusko, T.; Moldawer, L.; Reddy, S.T.; Guirgis, F.W. Lipid and Lipoprotein Dysregulation in Sepsis: Clinical and Mechanistic Insights into Chronic Critical Illness. J. Clin. Med. 2021, 10, 1693. [Google Scholar] [CrossRef] [PubMed]
- Van Wyngene, L.; Vanderhaeghen, T.; Timmermans, S.; Vandewalle, J.; Van Looveren, K.; Souffriau, J.; Wallaeys, C.; Eggermont, M.; Ernst, S.; Van Hamme, E.; et al. Hepatic PPARα function and lipid metabolic pathways are dysregulated in polymicrobial sepsis. EMBO Mol. Med. 2020, 12, e11319. [Google Scholar] [CrossRef]
- Vigneron, C.; Py, B.F.; Monneret, G.; Venet, F. The double sides of NLRP3 inflammasome activation in sepsis. Clin. Sci. 2023, 137, 333–351. [Google Scholar] [CrossRef]
- Law, S.H.; Chan, M.L.; Marathe, G.K.; Parveen, F.; Chen, C.H.; Ke, L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. International journal of molecular sciences 2019, 20, 1149. [Google Scholar] [CrossRef]
- Radu, C.G.; Yang, L.V.; Riedinger, M.; Au, M.; Witte, O.N. T cell chemotaxis to lysophosphatidylcholine through the G2A receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Drobnik, W.; Liebisch, G.; Audebert, F.X.; Frohlich, D.; Gluck, T.; Vogel, P.; Rothe, G.; Schmitz, G. Plasma ceramide and lysophosphatidylcholine inversely correlate with mortality in sepsis patients. J. Lipid Res. 2003, 44, 754–761. [Google Scholar] [CrossRef]
- Chiang, N.; Serhan, C.N. Specialized pro-resolving mediator network: An update on production and actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, G.; Wang, X.; Liu, D. Metabolic reprogramming consequences of sepsis: Adaptations and contradictions. Cell. Mol. Life Sci. CMLS 2022, 79, 456. [Google Scholar] [CrossRef]
- Chung, K.P.; Chen, G.Y.; Chuang, T.Y.; Huang, Y.T.; Chang, H.T.; Chen, Y.F.; Liu, W.L.; Chen, Y.J.; Hsu, C.L.; Huang, M.T.; et al. Increased Plasma Acetylcarnitine in Sepsis Is Associated with Multiple Organ Dysfunction and Mortality: A Multicenter Cohort Study. Crit. Care Med. 2019, 47, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, i1585. [Google Scholar] [CrossRef]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; McIntyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021, 47, 1181–1247. [Google Scholar] [CrossRef] [PubMed]
- van den Berghe, G.; Wouters, P.; Weekers, F.; Verwaest, C.; Bruyninckx, F.; Schetz, M.; Vlasselaers, D.; Ferdinande, P.; Lauwers, P.; Bouillon, R. Intensive insulin therapy in critically ill patients. N. Engl. J. Med. 2001, 345, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Langouche, L.; Vanhorebeek, I.; Vlasselaers, D.; Vander Perre, S.; Wouters, P.J.; Skogstrand, K.; Hansen, T.K.; Van den Berghe, G. Intensive insulin therapy protects the endothelium of critically ill patients. J. Clin. Investig. 2005, 115, 2277–2286. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, J.; Li, K.; Li, T.; Li, R.; Zhang, W.; Zhang, X.; Wang, Y.; Chen, M.; Shi, R.; et al. Metformin Alleviates Sepsis-Associated Myocardial Injury by Enhancing AMP-Activated Protein Kinase/Mammalian Target of Rapamycin Signaling Pathway-Mediated Autophagy. J. Cardiovasc. Pharmacol. 2023, 82, 308–317. [Google Scholar] [CrossRef]
- Maayah, Z.H.; Ferdaoussi, M.; Takahara, S.; Soni, S.; Dyck, J.R.B. Empagliflozin suppresses inflammation and protects against acute septic renal injury. Inflammopharmacology 2021, 29, 269–279. [Google Scholar] [CrossRef]
- Hu, L.; Chen, H.; Zhang, X.; Feng, Z.; Zhang, H.; Meng, Q. Rosiglitazone ameliorates radiation-induced intestinal inflammation in rats by inhibiting NLRP3 inflammasome and TNF-α production. J. Radiat. Res. 2020, 61, 842–850. [Google Scholar] [CrossRef]
- Kaplan, J.; Nowell, M.; Chima, R.; Zingarelli, B. Pioglitazone reduces inflammation through inhibition of NF-κB in polymicrobial sepsis. Innate Immun. 2014, 20, 519–528. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Wang, R.; Li, B.; Lam, S.M.; Shui, G. Integration of lipidomics and metabolomics for in-depth understanding of cellular mechanism and disease progression. J. Genet. Genom. 2020, 47, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Eckerle, M.; Ambroggio, L.; Puskarich, M.A.; Winston, B.; Jones, A.E.; Standiford, T.J.; Stringer, K.A. Metabolomics as a Driver in Advancing Precision Medicine in Sepsis. Pharmacotherapy 2017, 37, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Chouchane, O.; Schuurman, A.R.; Reijnders, T.D.Y.; Peters-Sengers, H.; Butler, J.M.; Uhel, F.; Schultz, M.J.; Bonten, M.J.; Cremer, O.L.; Calfee, C.S.; et al. The Plasma Lipidomic Landscape in Patients with Sepsis due to Community-acquired Pneumonia. Am. J. Respir. Crit. Care Med. 2024, 209, 973–986. [Google Scholar] [CrossRef]
- Jiang, M.; Li, L.; Jin, Y.; Lu, L.; Lu, Z.; Lv, W.; Wang, X.; Di, L.; Liu, Z. Derivative spectrophotometry-assisted determination of tryptophan metabolites emerges host and intestinal flora dysregulations during sepsis. Anal. Biochem. 2024, 694, 115605. [Google Scholar] [CrossRef]
- Shehata, A.H.; Anter, A.F.; Mohamed Naguib Abdel Hafez, S.; Rn Ibrahim, A.; Kamel, E.S.; Ahmed, A.F. Pioglitazone ameliorates sepsis-associated encephalopathy through SIRT1 signaling pathway. Int. Immunopharmacol. 2024, 139, 112757. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, G.; Zeng, Z. Effects of peroxisome proliferator-activated receptor-β/δ on sepsis induced acute lung injury. Chin. Med. J. 2014, 127, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zeng, Z.; Chen, A.; Lu, X.; Zhao, C.; Hu, C.; Zhou, L.; Liu, X.; Wang, X.; Hou, X.; et al. Comprehensive Strategy to Construct In-House Database for Accurate and Batch Identification of Small Molecular Metabolites. Anal. Chem. 2018, 90, 7635–7643. [Google Scholar] [CrossRef]
- Pecchillo Cimmino, T.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX Dependent ROS Generation and Cell Metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. Targeting sirtuin 1 to improve metabolism: All you need is NAD(+)? Pharmacol. Rev. 2012, 64, 166–187. [Google Scholar] [CrossRef]
- Rappou, E.; Jukarainen, S.; Rinnankoski-Tuikka, R.; Kaye, S.; Heinonen, S.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Saunavaara, V.; Rissanen, A.; et al. Weight Loss Is Associated With Increased NAD(+)/SIRT1 Expression But Reduced PARP Activity in White Adipose Tissue. J. Clin. Endocrinol. Metab. 2016, 101, 1263–1273. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.-y.; Wei, X.-c.; Wang, Q.; Yang, J.-y.; Hou, H.; Du, Z.-w.; Wu, X.-a. Effects of dibutyl phthalate on lipid metabolism in liver and hepatocytes based on PPARα/SREBP-1c/FAS/GPAT/AMPK signal pathway. Food Chem. Toxicol. 2021, 149, 112029. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Xie, X.; Wang, N.; Wang, Y.; Zhao, J.; Chen, F.; Qu, F.; Wen, W.; Miao, J.; Cui, H. Formononetin promotes fatty acid β-oxidation to treat non-alcoholic steatohepatitis through SIRT1/PGC-1α/PPARα pathway. Phytomedicine 2024, 124, 155285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Cai, X.F.; Ma, Y.L.; Lu, Q. Effect of rosiglitazone on myocardial injury in septic rats through NF-κB pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 452–460. [Google Scholar] [CrossRef]
- Yoo, S.H.; Abdelmegeed, M.A.; Song, B.-J. Activation of PPARα by Wy-14643 ameliorates systemic lipopolysaccharide-induced acute lung injury. Biochem. Biophys. Res. Commun. 2013, 436, 366–371. [Google Scholar] [CrossRef]
- Schaefer, M.B.; Pose, A.; Ott, J.; Hecker, M.; Behnk, A.; Schulz, R.; Weissmann, N.; Günther, A.; Seeger, W.; Mayer, K. Peroxisome proliferator-activated receptor-α reduces inflammation and vascular leakage in a murine model of acute lung injury. Eur. Respir. J. 2008, 32, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Orasanu, G.; Ziouzenkova, O.; Devchand, P.R.; Nehra, V.; Hamdy, O.; Horton, E.S.; Plutzky, J. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor-alpha-dependent manner in vitro and in vivo in mice. J. Am. Coll. Cardiol. 2008, 52, 869–881. [Google Scholar] [CrossRef]
- Hsiao, P.J.; Chiou, H.C.; Jiang, H.J.; Lee, M.Y.; Hsieh, T.J.; Kuo, K.K. Pioglitazone Enhances Cytosolic Lipolysis, β-oxidation and Autophagy to Ameliorate Hepatic Steatosis. Sci. Rep. 2017, 7, 9030. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Zingarelli, B.; Krallman, K.; Tang Girdwood, S.; Lagory, D.; Mizuno, T.; Fei, L.; Wong, H.R.; Vinks, A.A. Phase 1 safety and pharmacokinetic study on the use of pioglitazone in critically ill patients with sepsis: A randomized clinical trial. Intensive Care Med. 2018, 44, 2006–2008. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Zhang, J.-P.; Hu, J.-Y.; Huang, Y.-S. The potential regulatory roles of NAD+ and its metabolism in autophagy. Metabolism 2016, 65, 454–462. [Google Scholar] [CrossRef]
- Cros, C.; Margier, M.; Cannelle, H.; Charmetant, J.; Hulo, N.; Laganier, L.; Grozio, A.; Canault, M. Nicotinamide Mononucleotide Administration Triggers Macrophages Reprogramming and Alleviates Inflammation During Sepsis Induced by Experimental Peritonitis. Front. Mol. Biosci. 2022, 9, 895028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. TEM 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Krupa, M.M.; Pawlak, K. Kynurenine Pathway-An Underestimated Factor Modulating Innate Immunity in Sepsis-Induced Acute Kidney Injury? Cells 2022, 11, 2604. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Lv, T.; Zhang, T.; Feng, D.; Zhu, F.; Xu, Y.; Zhang, L.; Gu, L.; Guo, Z.; Ding, C.; et al. Interleukin-6 promotes skeletal muscle catabolism by activating tryptophan-indoleamine 2,3-dioxygenase 1-kynurenine pathway during intra-abdominal sepsis. J. Cachexia Sarcopenia Muscle 2023, 14, 1046–1059. [Google Scholar] [CrossRef]
- Cheng, S.; Chen, W.; Guo, Z.; Ding, C.; Zuo, R.; Liao, Q.; Liu, G. Paeonol alleviates ulcerative colitis by modulating PPAR-γ and nuclear factor-κB activation. Sci. Rep. 2024, 14, 18390. [Google Scholar] [CrossRef]
- Shan, D.; Li, J.L.; Wu, L.; Li, D.; Hurov, J.; Tobin, J.F.; Gimeno, R.E.; Cao, J. GPAT3 and GPAT4 are regulated by insulin-stimulated phosphorylation and play distinct roles in adipogenesis. J. Lipid Res. 2010, 51, 1971–1981. [Google Scholar] [CrossRef]
- Guan, T.; Zhou, X.; Zhou, W.; Lin, H. Regulatory T cell and macrophage crosstalk in acute lung injury: Future perspectives. Cell Death Discov. 2023, 9, 9. [Google Scholar] [CrossRef]
- Wang, Y.; Phelan, S.A.; Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Transgenic mice overexpressing peroxiredoxin 6 show increased resistance to lung injury in hyperoxia. Am. J. Respir. Cell Mol. Biol. 2006, 34, 481–486. [Google Scholar] [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Fiorenza, M.; Onslev, J.; Henríquez-Olguín, C.; Persson, K.W.; Hesselager, S.A.; Jensen, T.E.; Wojtaszewski, J.F.P.; Hostrup, M.; Bangsbo, J. Reducing the mitochondrial oxidative burden alleviates lipid-induced muscle insulin resistance in humans. Sci. Adv. 2024, 10, eadq4461. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, W.; Liao, B.; Yang, J. FFA-ROS-P53-mediated mitochondrial apoptosis contributes to reduction of osteoblastogenesis and bone mass in type 2 diabetes mellitus. Sci. Rep. 2015, 5, 12724. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Wang, X.; Liu, D. The potential mechanism of mitochondrial dysfunction in septic cardiomyopathy. J. Int. Med. Res. 2018, 46, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Zuev, S.M.; Kingsmore, S.F.; Gessler, D.D. Sepsis progression and outcome: A dynamical model. Theor. Biol. Med. Model. 2006, 3, 8. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Formula | m/z | RT (min) | CLP vs. Sham | Mode | Platform |
|---|---|---|---|---|---|---|
| Nicotinate * | C6H5NO2 | 125.0428 | 5.191 | up | Negative | LC-MS |
| L-Aspartate * | C4H7NO4 | 134.0448 | 1.03 | up | Negative | LC-MS |
| Hydroxyadipic acid * | C6H10O5 | 143.03385 | 1.386 | down | Negative | LC-MS |
| N2-gamma-Glutamylglutamine | C10H17N3O6 | 149.55524 | 6.81 | up | Positive | LC-MS |
| Orotic acid | C5H4N2O4 | 155.01058 | 1.423 | down | Negative | LC-MS |
| Quinolinate * | C7H5NO4 | 168.0291 | 1.437 | down | Negative | LC-MS |
| N-Acetylisoleucine | C8H15NO3 | 174.11229 | 6.78 | up | Positive | LC-MS |
| Glucosamine | C6H13NO5 | 180.08547 | 10.982 | up | Positive | LC-MS |
| Methoxytyrosine | C10H13NO4 | 192.06581 | 6.82 | up | Negative | LC-MS |
| Iduronic acid | C6H10O7 | 193.03461 | 1.425 | up | Negative | LC-MS |
| m-Methylhippuric acid | C10H11NO3 | 194.08097 | 6.811 | up | Positive | LC-MS |
| Propionylcarnitine * | C10H19NO4 | 198.11297 | 8.392 | up | Negative | LC-MS |
| methylene heptanoylglycine | C10H17NO3 | 200.12782 | 8.382 | up | Positive | LC-MS |
| L-Kynurenine * | C10H12N2O3 | 209.0928 | 6.493 | up | Positive | LC-MS |
| Naphthalenedicarboxylic acid | C12H8O4 | 215.03224 | 2.25 | down | Negative | LC-MS |
| 3-Hydroxy-L-Kynurenine * | C10H12N2O4 | 225.0872 | 7.119 | down | Positive | LC-MS |
| Methylacetoacetic acid | C5H8O3 | 231.08715 | 5.651 | down | Negative | LC-MS |
| Tetradecanedioic acid | C14H26O4 | 239.16498 | 11.746 | up | Negative | LC-MS |
| Leucyl-Aspartate | C10H18N2O5 | 247.12855 | 5.162 | up | Positive | LC-MS |
| Hexadecanedioic acid * | C16H30O4 | 267.19659 | 12.875 | up | Negative | LC-MS |
| L-Thyronine | C15H15NO4 | 274.1103 | 5.999 | up | Positive | LC-MS |
| Amino-1-MethylUracil | C5H7N3O2 | 283.11477 | 6.622 | up | Positive | LC-MS |
| Taurolithocholic acid 3-sulfate | C26H45NO8S2 | 293.64984 | 9.616 | up | Positive | LC-MS |
| N-octanoylglutamine | C13H24N2O4 | 295.16294 | 5.71 | up | Positive | LC-MS |
| Epoxyoctadecenoic acid | C18H32O3 | 295.22779 | 14.11 | up | Negative | LC-MS |
| N-Acetylneuraminic acid * | C11H19NO9 | 308.09873 | 1.463 | up | Negative | LC-MS |
| 2-hydroxy-dAMP | C10H14N5O7P | 346.0631 | 6.785 | up | Negative | LC-MS |
| Methyluric acid | C6H6N4O3 | 363.07568 | 6.853 | down | Negative | LC-MS |
| Eicosatrienoylethanolamide | C24H36O3 | 373.27321 | 9.817 | up | Positive | LC-MS |
| Linolenylcarnitine * | C25H43NO4 | 422.32587 | 11.679 | up | Positive | LC-MS |
| Arachidonoylcarnitine * | C27H46NO4 | 428.31662 | 16.531 | up | Negative | LC-MS |
| PI (22:5/18:0) | C49H85O13P | 468.2865 | 9.819 | up | Positive | LC-MS |
| Lignoceroylcarnitine * | C31H62NO4 | 512.46655 | 15.03 | down | Positive | LC-MS |
| LPE (22:1/0:0) | C27H54NO7P | 536.3343 | 10.026 | down | Positive | LC-MS |
| LysoPI (20:4/0:0) | C29H49O12P | 619.28882 | 13.46 | down | Negative | LC-MS |
| NAD+ | C21H27N7O14P2 | 663.3186 | 6.245 | down | Positive | LC-MS |
| L-Threonine * | C4H9NO3 | 219 | 15.34 | up | - | GC-MS |
| Oxalic acid * | C2H2O4 | 147 | 7.645 | up | - | GC-MS |
| L-Serine * | C3H7NO3 | 132 | 11.275 | up | - | GC-MS |
| Ethanolamine * | C2H7NO | 174 | 11.54 | up | - | GC-MS |
| Xylitol | C5H12O5 | 74 | 30.095 | down | - | GC-MS |
| Metabolite | Formula | m/z | RT (min) | CLP+Chi 10 vs. CLP | Mode | Platform |
|---|---|---|---|---|---|---|
| Nicotinate * | C6H5NO2 | 125.0428 | 5.191 | down | Negative | LC-MS |
| Methylhistamine | C6H11N3 | 126.10256 | 1.341 | down | Positive | LC-MS |
| Myristic acid * | C14H28O2 | 126.10257 | 1.224 | down | Positive | LC-MS |
| L-Aspartate * | C4H7NO4 | 134.0448 | 1.03 | up | Negative | LC-MS |
| Dihydroxy-2-methylpropionic acid | C4H8O4 | 143.03091 | 7.424 | down | Positive | LC-MS |
| Quinolinate * | C7H5NO4 | 168.0291 | 1.437 | up | Negative | LC-MS |
| L-Kynurenine * | C10H12N2O3 | 209.0928 | 6.493 | down | Positive | LC-MS |
| L-Glyceric acid * | C3H6O4 | 213.06197 | 12.119 | up | Positive | LC-MS |
| N-Acetyl-L-tyrosine * | C11H13NO4 | 222.08011 | 6.287 | up | Negative | LC-MS |
| 3-Hydroxy-L-Kynurenine * | C10H12N2O4 | 225.0872 | 6.119 | up | Positive | LC-MS |
| LysoPC (14:1/0:0) | C22H44NO7P | 244.63725 | 9.148 | up | Positive | LC-MS |
| L-Thyronine | C15H15NO4 | 274.1103 | 5.999 | down | Positive | LC-MS |
| 2-hydroxy-dAMP | C10H14N5O7P | 346.0631 | 6.785 | down | Negative | LC-MS |
| SM (d17:2/20:5-3OH(5,6,15)) | C42H73N2O9P | 389.24638 | 15.244 | down | Negative | LC-MS |
| Oxocholic acid | C24H38O5 | 405.26466 | 9.228 | up | Negative | LC-MS |
| LPE (22:1/0:0) | C27H54NO7P | 536.3343 | 10.026 | up | Positive | LC-MS |
| LysoPI (0:0/18:0) | C27H53O12P | 623.31586 | 5.563 | up | Positive | LC-MS |
| NAD+ | C21H27N7O14P2 | 663.3186 | 6.245 | up | Positive | LC-MS |
| Quillaic acid | C74H114O39 | 812.32325 | 6.797 | down | Negative | LC-MS |
| PI (18:1/16:0) | C43H81O13P | 835.53522 | 9.843 | up | Negative | LC-MS |
| PI (16:0/18:0) | C43H83O13P | 837.54931 | 9.845 | up | Negative | LC-MS |
| Xylitol | C5H12O5 | 74 | 30.095 | up | - | GC-MS |
| Oxalic acid * | C2H2O4 | 147 | 7.645 | down | - | GC-MS |
| Metabolite | Formula | m/z | RT (min) | CLP vs. Sham | CLP+Chi 10 vs. CLP | Mode | Platform |
|---|---|---|---|---|---|---|---|
| Quinolinate | C7H5NO4 | 168.0291 | 1.437 | down | up | Negative | LC-MS |
| L-Aspartate | C4H7NO4 | 134.0448 | 1.030 | up | up | Negative | LC-MS |
| 2-hydroxy-dAMP | C10H14N5O7P | 346.0631 | 6.785 | up | down | Negative | LC-MS |
| NAD+ | C21H27N7O14P2 | 663.3186 | 6.245 | down | up | Positive | LC-MS |
| L-Kynurenine | C10H12N2O3 | 209.0928 | 6.493 | up | down | Positive | LC-MS |
| Nicotinate | C6H5NO2 | 125.0428 | 5.191 | up | down | Negative | LC-MS |
| LPE (22:1/0:0) | C27H54NO7P | 536.3343 | 10.026 | down | up | Positive | LC-MS |
| L-Thyronine | C15H15NO4 | 274.1103 | 5.999 | up | down | Positive | LC-MS |
| 3-Hydroxy-L-Kynurenine | C10H12N2O4 | 225.0872 | 7.119 | down | up | Positive | LC-MS |
| Oxalic acid | C2H2O4 | 147 | 7.645 | down | up | - | GC-MS |
| Xylitol | C5H12O5 | 74 | 30.095 | up | down | - | GC-MS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, L.-L.; Cao, Y.-L.; Lu, Z.-C.; Wu, H.; Hu, S.-S.; Ye, B.-Q.; He, J.-Z.; Di, L.; Chen, X.-L.; Liu, Z.-C. Integrated Metabolomics and Lipidomics Analysis Reveals the Mechanism Behind the Action of Chiglitazar on the Protection Against Sepsis-Induced Acute Lung Injury. Metabolites 2025, 15, 290. https://doi.org/10.3390/metabo15050290
Lu L-L, Cao Y-L, Lu Z-C, Wu H, Hu S-S, Ye B-Q, He J-Z, Di L, Chen X-L, Liu Z-C. Integrated Metabolomics and Lipidomics Analysis Reveals the Mechanism Behind the Action of Chiglitazar on the Protection Against Sepsis-Induced Acute Lung Injury. Metabolites. 2025; 15(5):290. https://doi.org/10.3390/metabo15050290
Chicago/Turabian StyleLu, Liu-Liu, Yu-Li Cao, Zhen-Chen Lu, Han Wu, Shan-Song Hu, Bing-Qing Ye, Jin-Zhi He, Lei Di, Xu-Lin Chen, and Zhi-Cheng Liu. 2025. "Integrated Metabolomics and Lipidomics Analysis Reveals the Mechanism Behind the Action of Chiglitazar on the Protection Against Sepsis-Induced Acute Lung Injury" Metabolites 15, no. 5: 290. https://doi.org/10.3390/metabo15050290
APA StyleLu, L.-L., Cao, Y.-L., Lu, Z.-C., Wu, H., Hu, S.-S., Ye, B.-Q., He, J.-Z., Di, L., Chen, X.-L., & Liu, Z.-C. (2025). Integrated Metabolomics and Lipidomics Analysis Reveals the Mechanism Behind the Action of Chiglitazar on the Protection Against Sepsis-Induced Acute Lung Injury. Metabolites, 15(5), 290. https://doi.org/10.3390/metabo15050290