MALDI Mass Spectrometry Imaging for Visualizing In Situ Metabolism of Endogenous Metabolites and Dietary Phytochemicals

Abstract
:1. Introduction

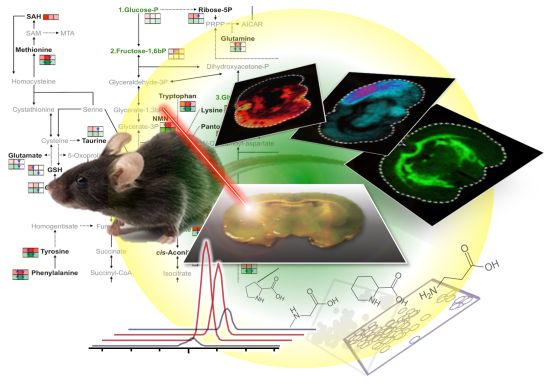
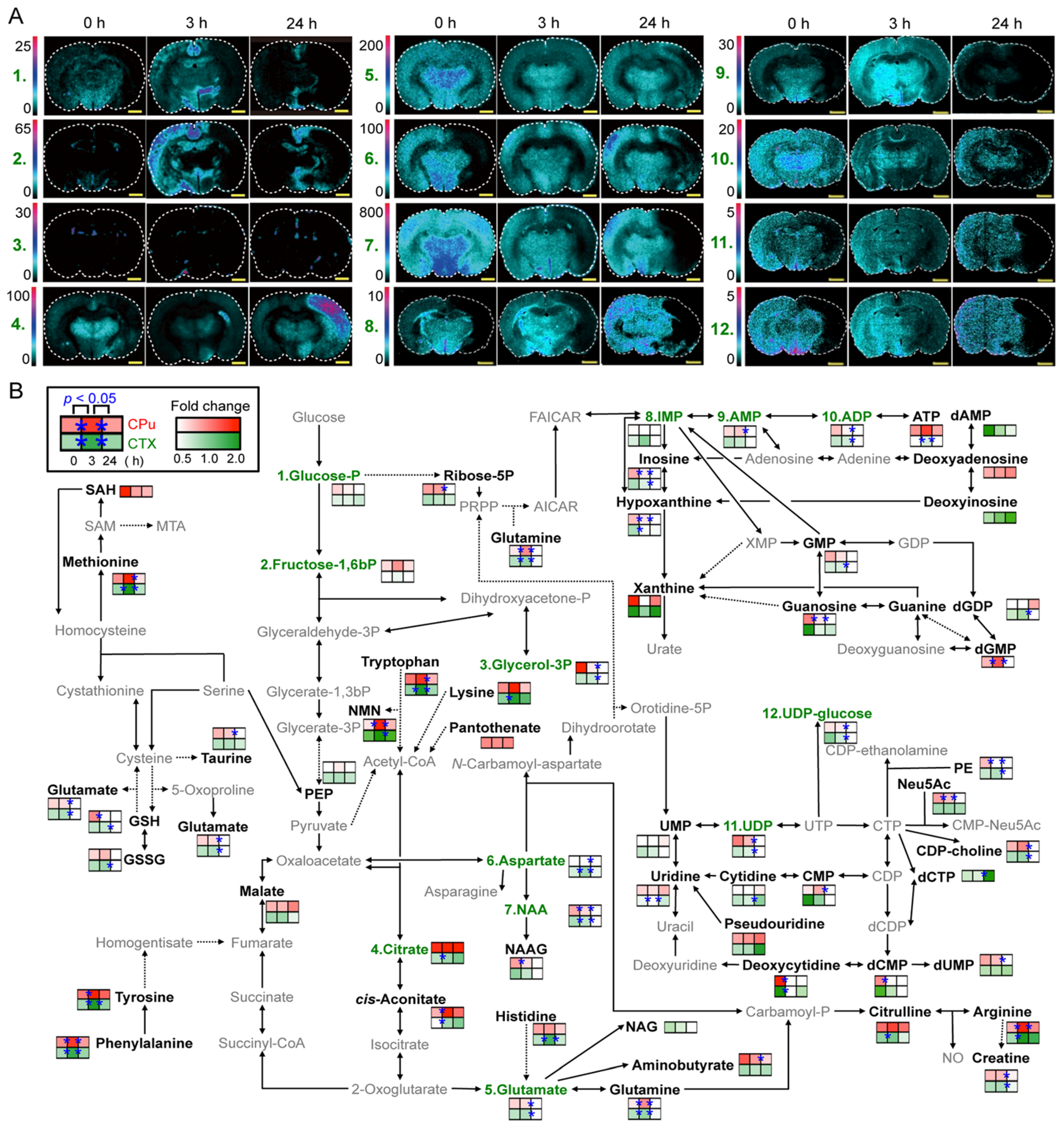
2. MALDI-MSI for Visualization of Endogenous Metabolite Distribution


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Analyte | Tissue | Species | Ref. |
|---|---|---|---|---|
| Animals | ||||
| DHB | Heme B | Liver | Human | [42] |
| Acetylcholine | Brain | Mouse | [53] | |
| 9-AA | Nucleotides, sugar phosphates | Brain | Rat | [23] |
| Nucleotides, sugar phosphates, organic acids, amino acids | Brain | Rat, Mouse | [52] | |
| 1,5-DAN | EGCG and its phase II metabolites | Liver, Kidney | Mouse | [54] |
| Edible plants | ||||
| DHB | γ-Oryzanol, α-tocopherol, phytic acid | Grain | Rice | [44] |
| GABA, amino acids, sugars | Fruit | Eggplant | [43] | |
| Glycoalkaloids | Tuber | Potato | [55] | |
| Anthocyanins | Fruit | Blueberry | [56] | |
| CHCA | Oligosaccharides | Stem | Wheat | [57] |
| Amino acids, sugars, sugar phosphates | Grain | Wheat | [58] | |
| Flavonoids, dihydrochalcones | Fruit | Apple | [59] | |
| 9-AA | Amino acids, sugars, sugar phosphates | Grain | Wheat | [58] |

3. Visualization of Dietary Phytochemical to Understand Its In Situ Metabolism

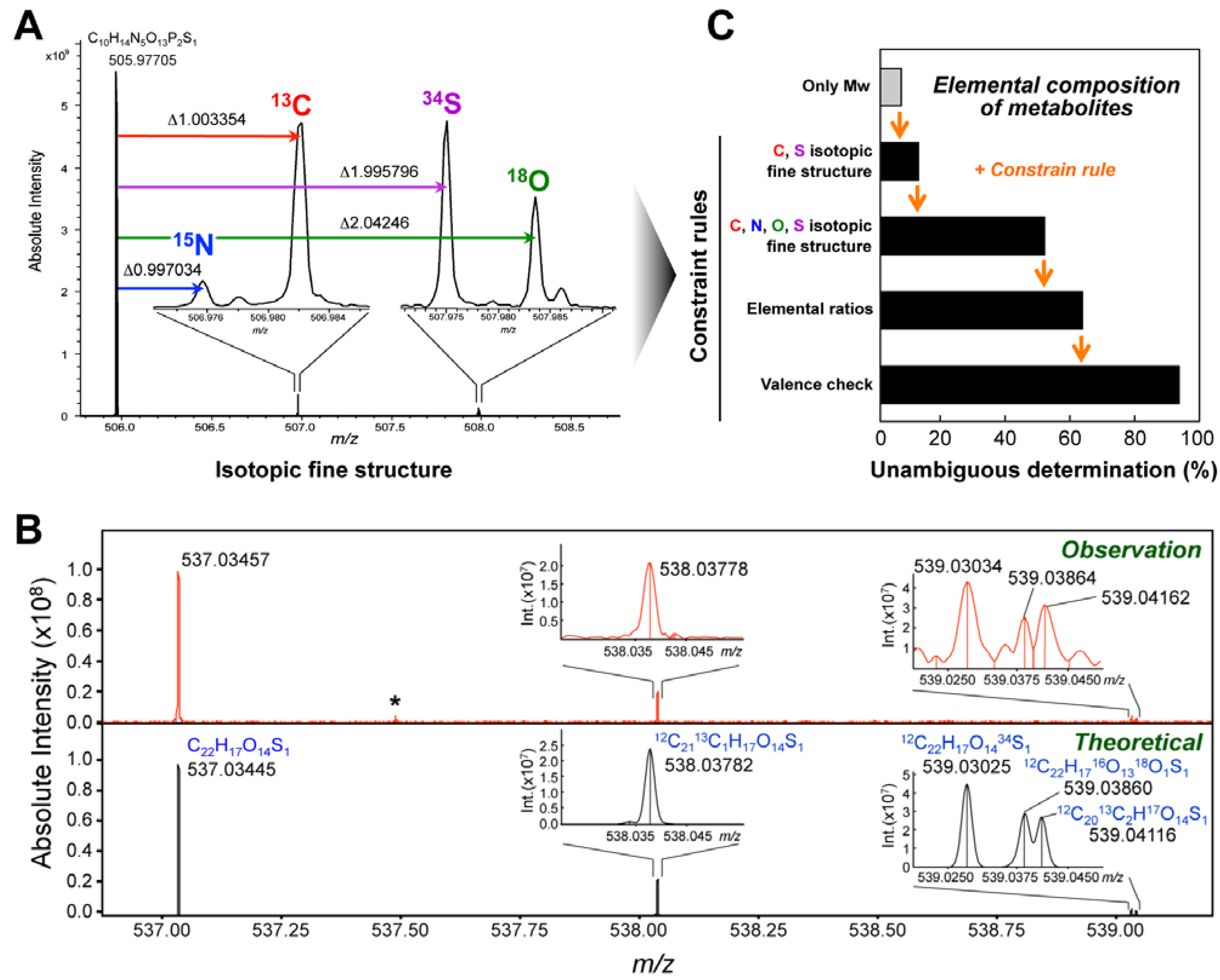
4. Metabolite Identification Strategy Using Ultrahigh-Resolution MS

5. The Rational Understanding of MALDI Ionization

6. Conclusion and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Villas-Boas, S.G.; Rasmussen, S.; Lane, G.A. Metabolomics or metabolite profiles? Trends Biotechnol. 2005, 23, 385–386. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Argmann, C.; Houten, S.M.; Canto, C.; Jeninga, E.H.; Andreux, P.A.; Thomas, C.; Doenlen, R.; Schoonjans, K.; Auwerx, J. The metabolic footprint of aging in mice. Sci. Rep. 2011, 1, 134. [Google Scholar]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef]
- Inoue, K.; Tsutsui, H.; Akatsu, H.; Hashizume, Y.; Matsukawa, N.; Yamamoto, T.; Toyo’oka, T. Metabolic profiling of Alzheimer’s disease brains. Sci. Rep. 2013, 3, 2364. [Google Scholar]
- Tritten, L.; Keiser, J.; Godejohann, M.; Utzinger, J.; Vargas, M.; Beckonert, O.; Holmes, E.; Saric, J. Metabolic profiling framework for discovery of candidate diagnostic markers of malaria. Sci. Rep. 2013, 3, 2769. [Google Scholar]
- Werner, E.; Croixmarie, V.; Umbdenstock, T.; Ezan, E.; Chaminade, P.; Tabet, J.C.; Junot, C. Mass spectrometry-based metabolomics: Accelerating the characterization of discriminating signals by combining statistical correlations and ultrahigh resolution. Anal. Chem. 2008, 80, 4918–4932. [Google Scholar] [CrossRef]
- Major, H.J.; Williams, R.; Wilson, A.J.; Wilson, I.D. A metabonomic analysis of plasma from Zucker rat strains using gas chromatography/mass spectrometry and pattern recognition. Rapid Commun. Mass Spectrom. 2006, 20, 3295–3302. [Google Scholar] [CrossRef]
- Pohjanen, E.; Thysell, E.; Jonsson, P.; Eklund, C.; Silfver, A.; Carlsson, I.B.; Lundgren, K.; Moritz, T.; Svensson, M.B.; Antti, H. A multivariate screening strategy for investigating metabolic effects of strenuous physical exercise in human serum. J. Proteome Res. 2007, 6, 2113–2120. [Google Scholar] [CrossRef]
- Judenhofer, M.S.; Wehrl, H.F.; Newport, D.F.; Catana, C.; Siegel, S.B.; Becker, M.; Thielscher, A.; Kneilling, M.; Lichy, M.P.; Eichner, M.; et al. Simultaneous PET-MRI: A new approach for functional and morphological imaging. Nat. Med. 2008, 14, 459–465. [Google Scholar] [CrossRef]
- Sugiura, Y.; Konishi, Y.; Zaima, N.; Kajihara, S.; Nakanishi, H.; Taguchi, R.; Setou, M. Visualization of the cell-selective distribution of PUFA-containing phosphatidylcholines in mouse brain by imaging mass spectrometry. J. Lipid Res. 2009, 50, 1776–1788. [Google Scholar] [CrossRef]
- Cornett, D.S.; Frappier, S.L.; Caprioli, R.M. MALDI-FTICR imaging mass spectrometry of drugs and metabolites in tissue. Anal. Chem. 2008, 80, 5648–5653. [Google Scholar] [CrossRef]
- Stoeckli, M.; Chaurand, P.; Hallahan, D.E.; Caprioli, R.M. Imaging mass spectrometry: A new technology for the analysis of protein expression in mammalian tissues. Nat. Med. 2001, 7, 493–496. [Google Scholar] [CrossRef]
- Caprioli, R.M.; Farmer, T.B.; Gile, J. Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Anal. Chem. 1997, 69, 4751–4760. [Google Scholar] [CrossRef]
- Chaurand, P.; Stoeckli, M.; Caprioli, R.M. Direct profiling of proteins in biological tissue sections by MALDI mass spectrometry. Anal. Chem. 1999, 71, 5263–5270. [Google Scholar] [CrossRef]
- Chaurand, P.; Norris, J.L.; Cornett, D.S.; Mobley, J.A.; Caprioli, R.M. New developments in profiling and imaging of proteins from tissue sections by MALDI mass spectrometry. J. ProteomeRes. 2006, 5, 2889–2900. [Google Scholar]
- Stoeckli, M.; Staab, D.; Staufenbiel, M.; Wiederhold, K.H.; Signor, L. Molecular imaging of amyloid beta peptides in mouse brain sections using mass spectrometry. Anal. Biochem. 2002, 311, 33–39. [Google Scholar] [CrossRef]
- Sugiura, Y.; Setou, M. Imaging mass spectrometry for visualization of drug and endogenous metabolite distribution: Toward in situ pharmacometabolomes. J. Neuroimmune Pharmacol. 2010, 5, 31–43. [Google Scholar] [CrossRef]
- Harada, T.; Yuba-Kubo, A.; Sugiura, Y.; Zaima, N.; Hayasaka, T.; Goto-Inoue, N.; Wakui, M.; Suematsu, M.; Takeshita, K.; Ogawa, K.; et al. Visualization of volatile substances in different organelles with an atmospheric-pressure mass microscope. Anal. Chem. 2009, 81, 9153–9157. [Google Scholar] [CrossRef]
- Trim, P.J.; Djidja, M.C.; Atkinson, S.J.; Oakes, K.; Cole, L.M.; Anderson, D.M.; Hart, P.J.; Francese, S.; Clench, M.R. Introduction of a 20 kHz Nd:YVO4 laser into a hybrid quadrupole time-of-flight mass spectrometer for MALDI-MS imaging. Anal. Bioanal. Chem. 2010, 397, 3409–3419. [Google Scholar] [CrossRef]
- Benabdellah, F.; Touboul, D.; Brunelle, A.; Laprevote, O. In situ primary metabolites localizationon a rat brain section by chemical mass spectrometry imaging. Anal. Chem. 2009, 81, 5557–5560. [Google Scholar] [CrossRef]
- Nilsson, A.; Fehniger, T.E.; Gustavsson, L.; Andersson, M.; Kenne, K.; Marko-Varga, G.; Andren, P.E. Fine mapping the spatial distribution and concentration of unlabeled drugs within tissue micro-compartments using imaging mass spectrometry. PLoS One 2010, 5, e11411. [Google Scholar]
- Baluya, D.L.; Garrett, T.J.; Yost, R.A. Automated MALDI matrix deposition method with inkjet printing for imaging mass spectrometry. Anal. Chem. 2007, 79, 6862–6867. [Google Scholar] [CrossRef]
- Aerni, H.R.; Cornett, D.S.; Caprioli, R.M. Automated acoustic matrix deposition for MALDI sample preparation. Anal. Chem. 2006, 78, 827–834. [Google Scholar] [CrossRef]
- Goodwin, R.J.; Mackay, C.L.; Nilsson, A.; Harrison, D.J.; Farde, L.; Andren, P.E.; Iverson, S.L. Qualitative and quantitative MALDI imaging of the positron emission tomography ligands raclopride (a D2 dopamine antagonist) and SCH 23390 (a D1 dopamine antagonist) in rat brain tissue sections using a solvent-free dry matrix application method. Anal. Chem. 2011, 83, 9694–9701. [Google Scholar]
- Goodwin, R.J.; Macintyre, L.; Watson, D.G.; Scullion, S.P.; Pitt, A.R. A solvent-free matrix application method for matrix-assisted laser desorption/ionization imaging of small molecules. Rapid Commun. Mass Spectrom. 2010, 24, 1682–1686. [Google Scholar] [CrossRef]
- Hankin, J.A.; Barkley, R.M.; Murphy, R.C. Sublimation as a method of matrix application for mass spectrometric imaging. J. Am. Soc. Mass Spectrom. 2007, 18, 1646–1652. [Google Scholar] [CrossRef]
- Thomas, A.; Charbonneau, J.L.; Fournaise, E.; Chaurand, P. Sublimation of new matrix candidates for high spatial resolution imaging mass spectrometry of lipids: Enhanced information in both positive and negative polarities after 1,5-diaminonapthalene deposition. Anal. Chem. 2012, 84, 2048–2054. [Google Scholar] [CrossRef]
- Yang, J.; Caprioli, R.M. Matrix sublimation/recrystallization for imaging proteins by mass spectrometry at high spatial resolution. Anal. Chem. 2011, 83, 5728–5734. [Google Scholar] [CrossRef]
- Murphy, R.C.; Hankin, J.A.; Barkley, R.M.; Zemski Berry, K.A. MALDI imaging of lipids after matrix sublimation/deposition. Biochim. Biophys. Acta 2011, 1811, 970–975. [Google Scholar]
- Bouschen, W.; Schulz, O.; Eikel, D.; Spengler, B. Matrix vapor deposition/recrystallization and dedicated spray preparation for high-resolution scanning microprobe matrix-assisted laser desorption/ionization imaging mass spectrometry (SMALDI-MS) of tissue and single cells. Rapid Commun. Mass Spectrom. 2010, 24, 355–364. [Google Scholar] [CrossRef]
- Tholey, A.; Heinzle, E. Ionic (liquid) matrices for matrix-assisted laser desorption/ionization mass spectrometry-applications and perspectives. Anal. Bioanal. Chem. 2006, 386, 24–37. [Google Scholar] [CrossRef]
- Meriaux, C.; Franck, J.; Wisztorski, M.; Salzet, M.; Fournier, I. Liquid ionic matrixes for MALDI mass spectrometry imaging of lipids. J. Proteomics 2010, 73, 1204–1218. [Google Scholar] [CrossRef]
- James, P.; Quadroni, M.; Carafoli, E.; Gonnet, G. Protein identification in DNA databases by peptide mass fingerprinting. Protein Sci. 1994, 3, 1347–1350. [Google Scholar] [CrossRef]
- Sun, G.; Yang, K.; Zhao, Z.; Guan, S.; Han, X.; Gross, R.W. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometric analysis of cellular glycerophospholipids enabled by multiplexed solvent dependent analyte-matrix interactions. Anal. Chem. 2008, 80, 7576–7585. [Google Scholar] [CrossRef]
- Goto-Inoue, N.; Yamada, K.; Inagaki, A.; Furuichi, Y.; Ogino, S.; Manabe, Y.; Setou, M.; Fujii, N.L. Lipidomics analysis revealed the phospholipid compositional changes in muscle by chronic exercise and high-fat diet. Sci. Rep. 2013, 3, 3267. [Google Scholar]
- Goto-Inoue, N.; Hayasaka, T.; Zaima, N.; Setou, M. Imaging mass spectrometry for lipidomics. Biochim. Biophys. Acta 2011, 1811, 961–969. [Google Scholar] [CrossRef]
- Murakami, M.; Nakatani, Y.; Atsumi, G.; Inoue, K.; Kudo, I. Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 1997, 17, 225–283. [Google Scholar] [CrossRef]
- Piomelli, D.; Astarita, G.; Rapaka, R. A neuroscientist's guide to lipidomics. Nat. Rev. Neurosci. 2007, 8, 743–754. [Google Scholar] [CrossRef]
- Shimma, S.; Setou, M. Mass microscopy to reveal distinct localization of heme B (m/z 616) in colon cancer liver metastasis. J. Mass Spectrom. Soc. Japan 2007, 55, 145–148. [Google Scholar] [CrossRef]
- Goto-Inoue, N.; Setou, M.; Zaima, N. Visualization of spatial distribution of gamma-aminobutyric acid in eggplant (Solanum melongena) by matrix-assisted laser desorption/ionization imaging mass spectrometry. Anal. Sci. 2010, 26, 821–825. [Google Scholar] [CrossRef]
- Zaima, N.; Goto-Inoue, N.; Hayasaka, T.; Setou, M. Application of imaging mass spectrometry for the analysis of Oryza sativa rice. Rapid Commun. Mass Spectrom. 2010, 24, 2723–2729. [Google Scholar] [CrossRef]
- Shroff, R.; Muck, A.; Svatos, A. Analysis of low molecular weight acids by negative mode matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 3295–3300. [Google Scholar] [CrossRef]
- Amantonico, A.; Oh, J.Y.; Sobek, J.; Heinemann, M.; Zenobi, R. Mass spectrometric method for analyzing metabolites in yeast with single cell sensitivity. Angew. Chem. Int. Ed. Engl. 2008, 47, 5382–5385. [Google Scholar] [CrossRef]
- Miura, D.; Fujimura, Y.; Tachibana, H.; Wariishi, H. Highly sensitive matrix-assisted laser desorption ionization-mass spectrometry for high-throughput metabolic profiling. Anal. Chem. 2010, 82, 498–504. [Google Scholar] [CrossRef]
- Yukihira, D.; Miura, D.; Saito, K.; Takahashi, K.; Wariishi, H. MALDI-MS-based high-throughput metabolite analysis for intracellular metabolic dynamics. Anal. Chem. 2010, 82, 4278–4282. [Google Scholar] [CrossRef]
- Sun, G.; Yang, K.; Zhao, Z.; Guan, S.; Han, X.; Gross, R.W. Shotgun metabolomics approach for the analysis of negatively charged water-soluble cellular metabolites from mouse heart tissue. Anal. Chem. 2007, 79, 6629–6640. [Google Scholar] [CrossRef]
- Edwards, J.L.; Kennedy, R.T. Metabolomic analysis of eukaryotic tissue and prokaryotes using negative mode MALDI time-of-flight mass spectrometry. Anal. Chem. 2005, 77, 2201–2209. [Google Scholar] [CrossRef]
- Shroff, R.; Vergara, F.; Muck, A.; Svatos, A.; Gershenzon, J. Nonuniform distribution of glucosinolates in Arabidopsis thaliana leaves has important consequences for plant defense. Proc. Natl. Acad. Sci. USA 2008, 105, 6196–6201. [Google Scholar] [CrossRef]
- Miura, D.; Fujimura, Y.; Yamato, M.; Hyodo, F.; Utsumi, H.; Tachibana, H.; Wariishi, H. Ultrahighly sensitive in situ metabolomic imaging for visualizing spatiotemporal metabolic behaviors. Anal. Chem. 2010, 82, 9789–9796. [Google Scholar] [CrossRef]
- Sugiura, Y.; Zaima, N.; Setou, M.; Ito, S.; Yao, I. Visualization of acetylcholine distribution in central nervous system tissue sections by tandem imaging mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1851–1861. [Google Scholar] [CrossRef]
- Kim, Y.H.; Fujimura, Y.; Hagihara, T.; Sasaki, M.; Yukihira, D.; Nagao, T.; Miura, D.; Yamaguchi, S.; Saito, K.; Tanaka, H.; et al. In situ label-free imaging for visualizing the biotransformation of a bioactive polyphenol. Sci. Rep. 2013, 3, 2805. [Google Scholar]
- Ha, M.; Kwak, J.H.; Kim, Y.; Zee, O.P. Direct analysis for the distribution of toxic glycoalkaloids in potato tuber tissue using matrix-assisted laser desorption/ionization mass spectrometric imaging. Food Chem. 2012, 133, 1155–1162. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Enomoto, H.; Moriyama, T.; Kawamura, Y.; Setou, M.; Zaima, N. Visualization of anthocyanin species in rabbiteye blueberry Vaccinium ashei by matrix-assisted laser desorption/ionization imaging mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1885–1895. [Google Scholar] [CrossRef]
- Robinson, S.; Warburton, K.; Seymour, M.; Clench, M.; Thomas-Oates, J. Localization of water-soluble carbohydrates in wheat stems using imaging matrix-assisted laser desorption ionization mass spectrometry. New Phytol. 2007, 173, 438–444. [Google Scholar] [CrossRef]
- Burrell, M.; Earnshaw, C.; Clench, M. Imaging Matrix Assisted Laser Desorption Ionization Mass Spectrometry: A technique to map plant metabolites within tissues at high spatial resolution. J. Exp. Bot. 2007, 58, 757–763. [Google Scholar] [CrossRef]
- Franceschi, P.; Dong, Y.; Strupat, K.; Vrhovsek, U.; Mattivi, F. Combining intensity correlation analysis and MALDI imaging to study the distribution of flavonols and dihydrochalcones in Golden Delicious apples. J. Exp. Bot. 2012, 63, 1123–1133. [Google Scholar] [CrossRef]
- Irie, M.; Fujimura, Y.; Yamato, M.; Miura, D.; Wariishi, H. Integrated MALDI-MS imaging and LC–MS techniques for visualizing spatiotemporal metabolomic dynamics in a rat stroke model. Metabolomics 2014, 10, 473–483. [Google Scholar] [CrossRef]
- Hattori, K.; Kajimura, M.; Hishiki, T.; Nakanishi, T.; Kubo, A.; Nagahata, Y.; Ohmura, M.; Yachie-Kinoshita, A.; Matsuura, T.; Morikawa, T.; et al. Paradoxical ATP elevation in ischemic penumbra revealed by quantitative imaging mass spectrometry. Antioxid. Redox Sign. 2010, 13, 1157–1167. [Google Scholar] [CrossRef]
- Sugiura, Y.; Honda, K.; Kajimura, M.; Suematsu, M. Visualization and quantification of cerebral metabolic fluxes of glucose in awake mice. Proteomics 2014, 14, 829–838. [Google Scholar]
- Goodwin, R.J. Sample preparation for mass spectrometry imaging: small mistakes can lead to big consequences. J. Proteomics 2012, 75, 4893–4911. [Google Scholar] [CrossRef]
- Kaletas, B.K.; van der Wiel, I.M.; Stauber, J.; Guzel, C.; Kros, J.M.; Luider, T.M.; Heeren, R.M. Sample preparation issues for tissue imaging by imaging MS. Proteomics 2009, 9, 2622–2633. [Google Scholar] [CrossRef]
- Shimma, S.; Takashima, Y.; Hashimoto, J.; Yonemori, K.; Tamura, K.; Hamada, A. Alternative two-step matrix application method for imaging mass spectrometry to avoid tissue shrinkage and improve ionization efficiency. J. Mass Spectrom. 2013, 48, 1285–1290. [Google Scholar] [CrossRef]
- Miura, D.; Fujimura, Y.; Wariishi, H. In situ metabolomic mass spectrometry imaging: Recent advances and difficulties. J. Proteomics 2012, 75, 5052–5060. [Google Scholar] [CrossRef]
- Prideaux, B.; Stoeckli, M. Mass spectrometry imaging for drug distribution studies. J. Proteomics 2012, 75, 4999–5013. [Google Scholar] [CrossRef]
- Solon, E.G.; Schweitzer, A.; Stoeckli, M.; Prideaux, B. Autoradiography, MALDI-MS, and SIMS-MS imaging in pharmaceutical discovery and development. AAPS J. 2010, 12, 11–26. [Google Scholar] [CrossRef]
- Riemann, B.; Schafers, K.P.; Schober, O.; Schafers, M. Small animal PET in preclinical studies: Opportunities and challenges. Q. J. Nucl. Med. Mol. Imag. 2008, 52, 215–221. [Google Scholar]
- Raskin, I.; Ribnicky, D.M.; Komarnytsky, S.; Ilic, N.; Poulev, A.; Borisjuk, N.; Brinker, A.; Moreno, D.A.; Ripoll, C.; Yakoby, N.; et al. Plants and human health in the twenty-first century. Trends Biotechnol. 2002, 20, 522–531. [Google Scholar] [CrossRef]
- Lee, Y.J.; Perdian, D.C.; Song, Z.; Yeung, E.S.; Nikolau, B.J. Use of mass spectrometry for imaging metabolites in plants. Plant J. 2012, 70, 81–95. [Google Scholar] [CrossRef]
- Kaspar, S.; Peukert, M.; Svatos, A.; Matros, A.; Mock, H.P. MALDI-imaging mass spectrometry—An emerging technique in plant biology. Proteomics 2011, 11, 1840–1850. [Google Scholar] [CrossRef]
- Matros, A.; Mock, H.P. Mass spectrometry based imaging techniques for spatially resolved analysis of molecules. Front. Plant Sci. 2013, 4, 89. [Google Scholar]
- Yoshimura, Y.; Zaima, N.; Moriyama, T.; Kawamura, Y. Different localization patterns of anthocyanin species in the pericarp of black rice revealed by imaging mass spectrometry. PLoS One 2012, 7, e31285. [Google Scholar]
- Chyu, K.Y.; Babbidge, S.M.; Zhao, X.; Dandillaya, R.; Rietveld, A.G.; Yano, J.; Dimayuga, P.; Cercek, B.; Shah, P.K. Differential effects of green tea-derived catechin on developing versus established atherosclerosis in apolipoprotein E-null mice. Circulation 2004, 109, 2448–2453. [Google Scholar] [CrossRef]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A receptor for green tea polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [CrossRef]
- Kumazoe, M.; Sugihara, K.; Tsukamoto, S.; Huang, Y.; Tsurudome, Y.; Suzuki, T.; Suemasu, Y.; Ueda, N.; Yamashita, S.; Kim, Y.; et al. 67-kDa laminin receptor increases cGMP to induce cancer-selective apoptosis. J. Clin. Invest. 2013, 123, 787–799. [Google Scholar]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Suzuki, T.; Kumazoe, M.; Kim, Y.; Yamashita, S.; Nakahara, K.; Tsukamoto, S.; Sasaki, M.; Hagihara, T.; Tsurudome, Y.; Huang, Y.; et al. Green tea extract containing a highly absorbent catechin prevents diet-induced lipid metabolism disorder. Sci. Rep. 2013, 3, 2749. [Google Scholar]
- Suganuma, M.; Okabe, S.; Oniyama, M.; Tada, Y.; Ito, H.; Fujiki, H. Wide distribution of [3H](-)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis 1998, 19, 1771–1776. [Google Scholar] [CrossRef]
- Yoshida, A.; Hirooka, Y.; Sugata, Y.; Nitta, M.; Manabe, T.; Ido, S.; Murakami, K.; Saha, R.K.; Suzuki, T.; Ohshima, M.; et al. Concise synthesis of catechin probes enabling analysis and imaging of EGCg. Chem. Commun. 2011, 47, 1794–1796. [Google Scholar] [CrossRef]
- Nakayama, M.; Shigemune, N.; Tsugukuni, T.; Tokuda, H.; Miyamoto, T. Difference of EGCg adhesion on cell surface between Staphylococcus aureus and Escherichia coli visualized by electron microscopy after novel indirect staining with cerium chloride. J. Microbiol. Methods 2011, 86, 97–103. [Google Scholar] [CrossRef]
- Sang, S.; Lambert, J.D.; Ho, C.T.; Yang, C.S. The chemistry and biotransformation of tea constituents. Pharmacol. Res. 2011, 64, 87–99. [Google Scholar] [CrossRef]
- Lambert, J.D.; Sang, S.; Yang, C.S. Biotransformation of green tea polyphenols and the biological activities of those metabolites. Mol. Pharm. 2007, 4, 819–825. [Google Scholar] [CrossRef]
- Yukihira, D.; Miura, D.; Fujimura, Y.; Umemura, Y.; Yamaguchi, S.; Funatsu, S.; Yamazaki, M.; Ohta, T.; Inoue, H.; Shindo, M.; et al. MALDI Efficiency of Metabolites Quantitatively Associated with their Structural Properties: A Quantitative Structure-Property Relationship (QSPR) Approach. J. Am. Soc. Mass Spectrom. 2013, 25, 1–5. [Google Scholar]
- Prideaux, B.; Dartois, V.; Staab, D.; Weiner, D.M.; Goh, A.; Via, L.E.; Barry, C.E., III; Stoeckli, M. High-sensitivity MALDI-MRM-MS imaging of moxifloxacin distribution in tuberculosis-infected rabbit lungs and granulomatous lesions. Anal. Chem. 2011, 83, 2112–2118. [Google Scholar] [CrossRef]
- Norris, J.L.; Caprioli, R.M. Analysis of tissue specimens by matrix-assisted laser desorption/ionization imaging mass spectrometry in biological and clinical research. Chem. Rev. 2013, 113, 2309–2342. [Google Scholar] [CrossRef]
- Chacon, A.; Zagol-Ikapitte, I.; Amarnath, V.; Reyzer, M.L.; Oates, J.A.; Caprioli, R.M.; Boutaud, O. On-tissue chemical derivatization of 3-methoxysalicylamine for MALDI-imaging mass spectrometry. J. Mass Spectrom. 2011, 46, 840–846. [Google Scholar] [CrossRef]
- Manier, M.L.; Reyzer, M.L.; Goh, A.; Dartois, V.; Via, L.E.; Barry, C.E., III; Caprioli, R.M. Reagent precoated targets for rapid in-tissue derivatization of the anti-tuberculosis drug isoniazid followed by MALDI imaging mass spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 1409–1419. [Google Scholar] [CrossRef]
- Louie, K.B.; Bowen, B.P.; McAlhany, S.; Huang, Y.; Price, J.C.; Mao, J.H.; Hellerstein, M.; Northen, T.R. Mass spectrometry imaging for in situ kinetic histochemistry. Sci. Rep. 2013, 3, 1656. [Google Scholar]
- Khatib-Shahidi, S.; Andersson, M.; Herman, J.L.; Gillespie, T.A.; Caprioli, R.M. Direct molecular analysis of whole-body animal tissue sections by imaging MALDI mass spectrometry. Anal. Chem. 2006, 78, 6448–6456. [Google Scholar] [CrossRef]
- Zavalin, A.; Todd, E.M.; Rawhouser, P.D.; Yang, J.; Norris, J.L.; Caprioli, R.M. Direct imaging of single cells and tissue at sub-cellular spatial resolution using transmission geometry MALDI MS. J. Mass Spectrom. 2012, 47, 1473–1481. [Google Scholar] [CrossRef]
- Yasunaga, M.; Furuta, M.; Ogata, K.; Koga, Y.; Yamamoto, Y.; Takigahira, M.; Matsumura, Y. The significance of microscopic mass spectrometry with high resolution in the visualisation of drug distribution. Sci. Rep. 2013, 3, 3050. [Google Scholar]
- Crecelius, A.C.; Cornett, D.S.; Caprioli, R.M.; Williams, B.; Dawant, B.M.; Bodenheimer, B. Three-dimensional visualization of protein expression in mouse brain structures using imaging mass spectrometry. J. Am. Soc. Mass Spectrom. 2005, 16, 1093–1099. [Google Scholar] [CrossRef]
- Seeley, E.H.; Caprioli, R.M. 3D imaging by mass spectrometry: A new frontier. Anal. Chem. 2012, 84, 2105–2110. [Google Scholar] [CrossRef]
- Liu, X.; Ide, J.L.; Norton, I.; Marchionni, M.A.; Ebling, M.C.; Wang, L.Y.; Davis, E.; Sauvageot, C.M.; Kesari, S.; Kellersberger, K.A.; et al. Molecular imaging of drug transit through the blood-brain barrier with MALDI mass spectrometry imaging. Sci. Rep. 2013, 3, 2859. [Google Scholar]
- Shimma, S.; Sugiura, Y.; Hayasaka, T.; Zaima, N.; Matsumoto, M.; Setou, M. Mass imaging and identification of biomolecules with MALDI-QIT-TOF-based system. Anal. Chem. 2008, 80, 878–885. [Google Scholar]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar]
- Houel, S.; Abernathy, R.; Renganathan, K.; Meyer-Arendt, K.; Ahn, N.G.; Old, W.M. Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. J. Proteome Res. 2010, 9, 4152–4160. [Google Scholar] [CrossRef]
- Niu, M.; Mao, X.; Ying, W.; Qin, W.; Zhang, Y.; Qian, X. Determination of monoisotopic masses of chimera spectra from high-resolution mass spectrometric data by use of isotopic peak intensity ratio modeling. Rapid Commun. Mass Spectrom. 2012, 26, 1875–1886. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics 2007, 8, 105. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics 2006, 7, 234. [Google Scholar] [CrossRef]
- Neumann, S.; Bocker, S. Computational mass spectrometry for metabolomics: Identification of metabolites and small molecules. Anal. Bioanal. Chem. 2010, 398, 2779–2788. [Google Scholar] [CrossRef]
- Kind, T.; Fiehn, O. Advances in structure elucidation of small molecules using mass spectrometry. Bioanal. Rev. 2010, 2, 23–60. [Google Scholar] [CrossRef]
- Giavalisco, P.; Hummel, J.; Lisec, J.; Inostroza, A.C.; Catchpole, G.; Willmitzer, L. High-resolution direct infusion-based mass spectrometry in combination with whole 13C metabolome isotope labeling allows unambiguous assignment of chemical sum formulas. Anal. Chem. 2008, 80, 9417–9425. [Google Scholar] [CrossRef]
- Rojas-Cherto, M.; Kasper, P.T.; Willighagen, E.L.; Vreeken, R.J.; Hankemeier, T.; Reijmers, T.H. Elemental composition determination based on MS(n). Bioinformatics 2011, 27, 2376–2383. [Google Scholar] [CrossRef]
- Matsuda, F.; Shinbo, Y.; Oikawa, A.; Hirai, M.Y.; Fiehn, O.; Kanaya, S.; Saito, K. Assessment of metabolome annotation quality: A method for evaluating the false discovery rate of elemental composition searches. PLoS One 2009, 4, e7490. [Google Scholar]
- Aharoni, A.; Ric de Vos, C.H.; Verhoeven, H.A.; Maliepaard, C.A.; Kruppa, G.; Bino, R.; Goodenowe, D.B. Nontargeted metabolome analysis by use of Fourier Transform Ion Cyclotron Mass Spectrometry. OMICS 2002, 6, 217–234. [Google Scholar] [CrossRef]
- Miura, D.; Tsuji, Y.; Takahashi, K.; Wariishi, H.; Saito, K. A strategy for the determination of the elemental composition by fourier transform ion cyclotron resonance mass spectrometry based on isotopic peak ratios. Anal. Chem. 2010, 82, 5887–5891. [Google Scholar] [CrossRef]
- Perdian, D.C.; Lee, Y.J. Imaging MS methodology for more chemical information in less data acquisition time utilizing a hybrid linear ion trap-orbitrap mass spectrometer. Anal. Chem. 2010, 82, 9393–9400. [Google Scholar] [CrossRef]
- Nagao, T.; Yukihira, D.; Fujimura, Y.; Saito, K.; Takahashi, K.; Miura, D.; Wariishi, H. Power of isotopic fine structure for unambiguous determination of metabolite elemental compositions: In silico evaluation and metabolomic application. Anal. Chim. Acta 2014, 813, 70–76. [Google Scholar] [CrossRef]
- Hamm, G.; Bonnel, D.; Legouffe, R.; Pamelard, F.; Delbos, J.M.; Bouzom, F.; Stauber, J. Quantitative mass spectrometry imaging of propranolol and olanzapine using tissue extinction calculation as normalization factor. J. Proteomics 2012, 75, 4952–4961. [Google Scholar] [CrossRef]
- Kallback, P.; Shariatgorji, M.; Nilsson, A.; Andren, P.E. Novel mass spectrometry imaging software assisting labeled normalization and quantitation of drugs and neuropeptides directly in tissue sections. J. Proteomics 2012, 75, 4941–4951. [Google Scholar] [CrossRef]
- Knochenmuss, R. Ion formation mechanisms in UV-MALDI. Analyst 2006, 131, 966–986. [Google Scholar] [CrossRef]
- Cerruti, C.D.; Benabdellah, F.; Laprevote, O.; Touboul, D.; Brunelle, A. MALDI imaging and structural analysis of rat brain lipid negative ions with 9-aminoacridine matrix. Anal. Chem. 2012, 84, 2164–2171. [Google Scholar] [CrossRef]
- Eibisch, M.; Schiller, J. Sphingomyelin is more sensitively detectable as a negative ion than phosphatidylcholine: A matrix-assisted laser desorption/ionization time-of-flight mass spectrometric study using 9-aminoacridine (9-AA) as matrix. Rapid Commun. Mass Spectrom. 2011, 25, 1100–1106. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Shroff, R.; Rulisek, L.; Doubsky, J.; Svatos, A. Acid-base-driven matrix-assisted mass spectrometry for targeted metabolomics. Proc. Natl. Acad. Sci. USA 2009, 106, 10092–10096. [Google Scholar] [CrossRef]
- Jaskolla, T.W.; Lehmann, W.D.; Karas, M. 4-Chloro-alpha-cyanocinnamic acid is an advanced, rationally designed MALDI matrix. Proc. Natl. Acad. Sci. USA 2008, 105, 12200–12205. [Google Scholar] [CrossRef]
- Wang, X.; Han, J.; Pan, J.; Borchers, C.H. Comprehensive imaging of porcine adrenal gland lipids by MALDI-FTMS using quercetin as a matrix. Anal. Chem. 2014, 86, 638–646. [Google Scholar] [CrossRef]
- Wang, X.; Han, J.; Chou, A.; Yang, J.; Pan, J.; Borchers, C.H. Hydroxyflavones as a new family of matrices for MALDI tissue imaging. Anal. Chem. 2013, 85, 7566–7573. [Google Scholar] [CrossRef]
- Annesley, T.M. Ion suppression in mass spectrometry. Clin. Chem. 2003, 49, 1041–1044. [Google Scholar] [CrossRef]
- Prideaux, B.; Staab, D.; Stoeckli, M. Applications of MALDI-MSI to pharmaceutical research. Methods Mol. Biol. 2010, 656, 405–413. [Google Scholar] [CrossRef]
- Li, Y.; Shrestha, B.; Vertes, A. Atmospheric pressure infrared MALDI imaging mass spectrometry for plant metabolomics. Anal. Chem. 2008, 80, 407–420. [Google Scholar] [CrossRef]
- Li, Y.; Shrestha, B.; Vertes, A. Atmospheric pressure molecular imaging by infrared MALDI mass spectrometry. Anal. Chem. 2007, 79, 523–532. [Google Scholar] [CrossRef]
- Taira, S.; Sugiura, Y.; Moritake, S.; Shimma, S.; Ichiyanagi, Y.; Setou, M. Nanoparticle-assisted laser desorption/ionization based mass imaging with cellular resolution. Anal. Chem. 2008, 80, 4761–4766. [Google Scholar] [CrossRef]
- Schober, Y.; Guenther, S.; Spengler, B.; Rompp, A. Single cell matrix-assisted laser desorption/ionization mass spectrometry imaging. Anal. Chem. 2012, 84, 6293–6297. [Google Scholar] [CrossRef]
- Deininger, S.O.; Cornett, D.S.; Paape, R.; Becker, M.; Pineau, C.; Rauser, S.; Walch, A.; Wolski, E. Normalization in MALDI-TOF imaging datasets of proteins: Practical considerations. Anal. Bioanal. Chem. 2011, 401, 167–181. [Google Scholar] [CrossRef]
- Garrett, T.J.; Yost, R.A. Analysis of intact tissue by intermediate-pressure MALDI on a linear ion trap mass spectrometer. Anal. Chem. 2006, 78, 2465–2469. [Google Scholar] [CrossRef]
- Hsieh, Y.; Casale, R.; Fukuda, E.; Chen, J.; Knemeyer, I.; Wingate, J.; Morrison, R.; Korfmacher, W. Matrix-assisted laser desorption/ionization imaging mass spectrometry for direct measurement of clozapine in rat brain tissue. Rapid Commun. Mass Spectrom. 2006, 20, 965–972. [Google Scholar] [CrossRef]
- Jackson, S.N.; Ugarov, M.; Egan, T.; Post, J.D.; Langlais, D.; Albert Schultz, J.; Woods, A.S. MALDI-ion mobility-TOFMS imaging of lipids in rat brain tissue. J. Mass Spectrom. 2007, 42, 1093–1098. [Google Scholar] [CrossRef]
- McLean, J.A.; Ridenour, W.B.; Caprioli, R.M. Profiling and imaging of tissues by imaging ion mobility-mass spectrometry. J. Mass Spectrom. 2007, 42, 1099–1105. [Google Scholar] [CrossRef]
- Trim, P.J.; Henson, C.M.; Avery, J.L.; McEwen, A.; Snel, M.F.; Claude, E.; Marshall, P.S.; West, A.; Princivalle, A.P.; Clench, M.R. Matrix-assisted laser desorption/ionization-ion mobility separation-mass spectrometry imaging of vinblastine in whole body tissue sections. Anal. Chem. 2008, 80, 8628–8634. [Google Scholar] [CrossRef]
- Liu, Q.; Xiao, Y.; Pagan-Miranda, C.; Chiu, Y.M.; He, L. Metabolite imaging using matrix-enhanced surface-assisted laser desorption/ionization mass spectrometry (ME-SALDI-MS). J. Am. Soc. Mass Spectrom. 2009, 20, 80–88. [Google Scholar] [CrossRef]
- Liu, Q.; Guo, Z.; He, L. Mass spectrometry imaging of small molecules using desorption/ionization on silicon. Anal. Chem. 2007, 79, 3535–3541. [Google Scholar] [CrossRef]
- Northen, T.R.; Yanes, O.; Northen, M.T.; Marrinucci, D.; Uritboonthai, W.; Apon, J.; Golledge, S.L.; Nordstrom, A.; Siuzdak, G. Clathrate nanostructures for mass spectrometry. Nature 2007, 449, 1033–1036. [Google Scholar] [CrossRef]
- Sun, N.; Walch, A. Qualitative and quantitative mass spectrometry imaging of drugs and metabolites in tissue at therapeutic levels. Histochem. Cell. Biol. 2013, 140, 93–104. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fujimura, Y.; Miura, D. MALDI Mass Spectrometry Imaging for Visualizing In Situ Metabolism of Endogenous Metabolites and Dietary Phytochemicals. Metabolites 2014, 4, 319-346. https://doi.org/10.3390/metabo4020319
Fujimura Y, Miura D. MALDI Mass Spectrometry Imaging for Visualizing In Situ Metabolism of Endogenous Metabolites and Dietary Phytochemicals. Metabolites. 2014; 4(2):319-346. https://doi.org/10.3390/metabo4020319
Chicago/Turabian StyleFujimura, Yoshinori, and Daisuke Miura. 2014. "MALDI Mass Spectrometry Imaging for Visualizing In Situ Metabolism of Endogenous Metabolites and Dietary Phytochemicals" Metabolites 4, no. 2: 319-346. https://doi.org/10.3390/metabo4020319
APA StyleFujimura, Y., & Miura, D. (2014). MALDI Mass Spectrometry Imaging for Visualizing In Situ Metabolism of Endogenous Metabolites and Dietary Phytochemicals. Metabolites, 4(2), 319-346. https://doi.org/10.3390/metabo4020319