Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma

 , ,
, , _Verhoeven-Duif.png)

Abstract
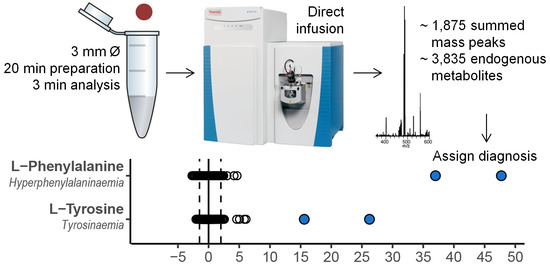
:
1. Introduction
2. Results
2.1. Reproducibility Assessment
2.2. Evaluation of the Clinical Value of the Method
2.3. Direct-Infusion Based Metabolomics in Metabolic Diagnostics
3. Discussion
4. Methods
4.1. Sample Collection
4.2. Patient Inclusion and Sample Selection
4.3. Sample Preparation
4.4. DI-HRMS Analysis
4.5. Data Processing
4.6. Data Analysis
4.7. Evaluation of the Clinical Value of the Method
4.8. Reproducibility Assessment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Janeckova, H.; Hron, K.; Wojtowicz, P.; Hlídková, E.; Barešová, A.; Friedecký, D.; Zídková, L.; Hornik, P.; Behúlová, D.; Procházková, D.; et al. Targeted metabolomic analysis of plasma samples for the diagnosis of inherited metabolic disorders. J. Chromatogr. A 2012, 1226, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.J.; Eggington, M.; Kahler, S.G. Comprehensive screening of urine samples for inborn errors of metabolism by electrospray tandem mass spectrometry. Clin. Chem. 2002, 48, 1970–1980. [Google Scholar] [PubMed]
- Jacob, M.; Malkawi, A.; Albast, N.; Al Bougha, S.; Lopata, A.; Dasouki, M.; Abdel Rahman, A.M. A targeted metabolomics approach for clinical diagnosis of inborn errors of metabolism. Analyt. Chim. Acta 2018, 1025, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Kurko, J.; Tringham, M.; Tanner, L.; Näntö-Salonen, K.; Vähä-Mäkilä, M.; Nygren, H.; Pöhö, P.; Lietzen, N.; Mattila, I.; Olkku, A.; et al. Imbalance of plasma amino acids, metabolites and lipids in patients with lysinuric protein intolerance. Metabolism 2016, 65, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Dercksen, M.; Koekemoer, G.; Duran, M.; Wanders, R.J.A.; Mienie, L.J.; Reinecke, C.J. Organic acid profile of isovaleric acidemia: A comprehensive metabolomics approach. Metabolomics 2013, 9, 765–777. [Google Scholar] [CrossRef]
- Smuts, I.; van der Westhuizen, F.H.; Louw, R.; Mienie, L.J.; Engelke, U.F.H.; Wevers, R.A.; Mason, S.; Koekemoer, G.; Reinecke, C.J. Disclosure of a putative biosignature for respiratory chain disorders through a metabolomics approach. Metabolomics 2013, 9, 379–391. [Google Scholar] [CrossRef]
- Najdekr, L.; Gardlo, A.; Madrova, L.; Friedecký, D.; Janečková, H.; Correa, E.S.; Goodacre, R.; Adam, T. Oxidized phosphatidylcholines suggest oxidative stress in patients with medium-chain acyl-CoA dehydrogenase deficiency. Talanta 2015, 139, 62–66. [Google Scholar] [CrossRef]
- Tebani, A.; Schmitz-Afonso, I.; Abily-Donval, L.; Héron, B.; Piraud, M.; Ausseil, J.; Brassier, A.; De Lonlay, P.; Zerimech, F.; Vaz, F.M.; et al. Urinary metabolic phenotyping of mucopolysaccharidosis type I combining untargeted and targeted strategies with data modeling. Clin. Chim. Acta 2017, 475, 7–14. [Google Scholar] [CrossRef]
- Wangler, M.F.; Huber, L.; Donti, T.R.; Ventura, M.J.; Miller, M.J.; Braverman, N.; Gawron, K.; Bose, M.; Moser, A.B.; Jones, R.O.; et al. A metabolomics map of Zellweger spectrum disorders reveals novel disease biomarkers. Genet. Med. 2018, 20, 1274–1283. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Gangoiti, J.A.; Barshop, B.A.; Siuzdak, G. Metabolomics identifies perturbations in human disorders of propionate metabolism. Clin. Chem. 2007, 53, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Denes, J.; Szabo, E.; Robinette, S.L.; Szatmári, I.; Szőnyi, L.; Kreuder, J.G.; Rauterberg, E.W.; Takáts, Z. Metabonomics of newborn screening dried blood spot samples: A novel approach in the screening and diagnostics of inborn errors of metabolism. Anal. Chem. 2012, 84, 10113–10120. [Google Scholar] [CrossRef] [PubMed]
- Peretz, H.; Watson, D.G.; Blackburn, G.; Zhang, T.; Lagziel, A.; Shtauber-Naamati, M.; Morad, T.; Keren-Tardai, E.; Greenshpun, V.; Usher, S.; et al. Urine metabolomics reveals novel physiologic functions of human aldehyde oxidase and provides biomarkers for typing xanthinuria. Metabolomics 2012, 8, 951–959. [Google Scholar] [CrossRef]
- Atwal, P.S.; Donti, T.R.; Cardon, A.L.; Bacino, C.A.; Sun, Q.; Emrick, L.; Reid Sutton, V.; Elsea, S.H. Aromatic L-amino acid decarboxylase deficiency diagnosed by clinical metabolomics profiling of plasma. Mol. Genet. Metab. 2015, 115, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Venter, L.; Lindeque, Z.; Jansen van Rensburg, P.; van der Westhuizen, F.; Smuts, I.; Louw, R. Untargeted urine metabolomics reveals a biosignature for muscle respiratory chain deficiencies. Metabolomics 2015, 11, 111–121. [Google Scholar] [CrossRef]
- Abela, L.; Simmons, L.; Steindl, K.; Schmitt, B.; Mastrangelo, M.; Joset, P.; Papuc, M.; Sticht, H.; Baumer, A.; Crowther, L.M.; et al. N(8)-acetylspermidine as a potential plasma biomarker for Snyder-Robinson syndrome identified by clinical metabolomics. J. Inherit. Metab. Dis. 2016, 39, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Donti, T.R.; Cappuccio, G.; Hubert, L.; Neira, J.; Atwal, P.S.; Miller, M.J.; Cardon, A.L.; Sutton, V.R.; Porter, B.E.; Baumer, F.M.; et al. Diagnosis of adenylosuccinate lyase deficiency by metabolomic profiling in plasma reveals a phenotypic spectrum. Mol. Genet. Metab. Rep. 2016, 8, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Bostwick, B.L.; Kennedy, A.D.; Donti, T.R.; Sun, Q.; Sutton, V.R.; Elsea, S.H. Chronic oral L-carnitine supplementation drives marked plasma TMAO elevations in patients with organic acidemias despite dietary meat restrictions. JIMD Rep. 2016, 30, 39–44. [Google Scholar]
- Abela, L.; Spiegel, R.; Crowther, L.M.; Klein, A.; Steindl, K.; Papuc, S.M.; Joset, P.; Zehavi, Y.; Rauch, A.; Plecko, B.; et al. Plasma metabolomics reveals a diagnostic metabolic fingerprint for mitochondrial aconitase (ACO2) deficiency. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Glinton, K.E.; Benke, P.J.; Lines, M.A.; Geraghty, M.T.; Chakraborty, P.; Al-Dirbashi, O.Y.; Jiang, Y.; Kennedy, A.D.; Grotewiel, M.S.; Suttona, V.R.; et al. Disturbed phospholipid metabolism in serine biosynthesis defects revealed by metabolomic profiling. Mol. Genet. Metab. 2018, 123, 309–316. [Google Scholar] [CrossRef]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sain-van der Velden, M.G.M.; van der Ham, M.; Gerrits, J.; Prinsen, H.C.M.T.; Willemsen, M.; Pras-Raves, M.L.; Jans, J.J.; Verhoeven-Duif, N.M. Quantification of metabolites in dried blood spots by direct infusion high resolution mass spectrometry. Anal. Chim. Acta 2017, 979, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, V.; Van Berkel, G.J. Fully automated liquid extraction-based surface sampling and ionization using a chip-based robotic nanoelectrospray platform. J. Mass Spectrom. 2010, 45, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, J.A.; Broadhurst, D.I.; Davidson, R.L.; Viant, M.R. Characterising and correcting batch variation in an automated direct infusion mass spectrometry (DIMS) metabolomics workflow. Anal. Bioanal. Chem. 2013, 405, 5147–5157. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J.L. Using direct infusion mass spectrometry for serum metabolomics in Alzheimer’s disease. Anal. Bioanal. Chem. 2014, 406, 7137–7148. [Google Scholar] [CrossRef] [PubMed]
- Lokhov, P.G.; Trifonova, O.P.; Maslov, D.L.; Balashova, E.E.; Archakov, A.I.; Shestakova, E.A.; Shestakova, M.V.; Dedov, I.I. Diagnosing impaired glucose intolerance using direct infusion mass spectrometry of blood plasma. PLoS ONE 2014, 9, e105343. [Google Scholar] [CrossRef] [PubMed]
- Koulman, A.; Prentice, P.; Wong, M.C.Y.; Matthews, L.; Bond, N.J.; Eiden, M.; Griffin, J.L.; Dunger, D.B. The development and validation of a fast and robust dried blood spot based lipid profiling method to study infant metabolism. Metabolomics 2014, 10, 1018–1025. [Google Scholar] [CrossRef] [Green Version]
- Anand, S.; Barnes, J.M.; Young, S.A.; Garcia, D.M.; Tolley, H.D.; Kauwe, J.S.K.; Graves, S.W. Discovery and confirmation of diagnostic serum lipid biomarkers for Alzheimer’s disease using direct infusion mass spectrometry. J. Alzheimers Dis. 2017, 59, 277–290. [Google Scholar] [CrossRef]
- Anand, S.; Young, S.; Esplin, M.S.; Peaden, B.; Tolley, H.D.; Porter, T.F.; Varner, M.W.; D’Alton, M.E.; Jackson, B.J.; Graves, S.W. Detection and confirmation of serum lipid biomarkers for preeclampsia using direct infusion mass spectrometry. J. Lipid Res. 2017, 57, 687–696. [Google Scholar] [CrossRef]
- Ramos, R.J.; Pras-Raves, M.L.; Gerrits, J.; van der Ham, M.; Willemsen, M.; Prinsen, H.; Burgering, B.; Jans, J.J.; Verhoeven-Duif, N.M. Vitamin B6 is essential for serine de novo biosynthesis. J. Inherit. Metab. Dis. 2017, 40, 883–891. [Google Scholar] [CrossRef]
- Sain-van der Velden, M.G.M.; Diekman, E.F.; Jans, J.J.; van der Ham, M.; Prinsen, B.H.; Visser, G.; Verhoeven-Duif, N.M. Differences between acylcarnitine profiles in plasma and bloodspots. Mol. Genet. Metabol. 2013, 110, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Primassin, S.; Spiekerkoetter, U. ESI-MS/MS measurement of free carnitine and its precursor γ-butyrobetaine in plasma and dried blood spots from patients with organic acidurias and fatty acid oxidation disorders. Mol. Genet. Metabol. 2010, 101, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Heiner-Fokkema, M.R.; Vaz, F.M.; Maatman, R.; Kluijtmans, L.A.J.; van Spronsen, F.J.; Reijngoud, D.J. Reliable diagnosis of carnitine palmitoyltransferase type IA deficiency by analysis of plasma acylcarnitine profiles. JIMD Rep. 2016, 32, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, 801–807. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DBS | Plasma | ||||||
|---|---|---|---|---|---|---|---|
| Batch 1 | Batch 2 | Batch 3 | Batch 4 | Batch 1 | Batch 2 | Batch 3 | |
| Mass peak fitting | 185,661 | 176,934 | 197,681 | 190,172 | 192,198 | 177,879 | 185,642 |
| Mass peak annotation | 59,543 | 56,250 | 63,360 | 60,979 | 62,503 | 58,212 | 60,450 |
| Adduct summation | 6580 | 6625 | 6598 | 6611 | |||
| Endogenous mass peaks * | 1874 | 1885 | 1874 | 1875 | 1875 | 1867 | 1868 |
| Endogenous metabolite annotations * | 3822 | 3863 | 3826 | 3839 | 3832 | 3847 | 3817 |
| DBS | Plasma | ||||||
|---|---|---|---|---|---|---|---|
| Batch 1 | Batch 2 | Batch 3 | Batch 4 | Batch 1 | Batch 2 | Batch 3 | |
| 15N;2−13C-glycine | 0.23 | 0.16 | 0.18 | 0.24 | 0.22 | 0.21 | 0.79 |
| 2H4-alanine | 0.20 | 0.14 | 0.16 | 0.20 | 0.20 | 0.21 | 0.19 |
| 2H3-leucine | 0.18 | 0.14 | 0.15 | 0.18 | 0.60 | 0.55 | 0.50 |
| 2H3-methionine | 0.31 | 0.30 | 0.36 | 0.39 | 1.70 | 0.22 | 0.20 |
| 13C6-phenylalanine | 0.19 | 0.16 | 0.14 | 0.18 | 0.21 | 0.20 | 0.19 |
| 13C6-tyrosine | 0.19 | 0.17 | 0.16 | 0.20 | 0.22 | 0.21 | 0.18 |
| 2H3-aspartate | 0.24 | 0.22 | 0.22 | 0.25 | 0.23 | 0.24 | 0.26 |
| 2H3-glutamate | 0.17 | 0.15 | 0.14 | 0.18 | 0.20 | 0.21 | 0.15 |
| 2H2-ornithine | 0.21 | 0.19 | 0.17 | 0.21 | 0.14 | 0.17 | 0.12 |
| 2H2-citrulline | 0.16 | 0.16 | 0.14 | 0.18 | 0.18 | 0.19 | 0.14 |
| 2H4;13C-arginine | 0.21 | 0.17 | 0.16 | 0.20 | 0.17 | 0.18 | 0.16 |
| 2H8-valine | 0.18 | 0.14 | 0.15 | 0.18 | 0.20 | 0.19 | 0.18 |
| 2H9-carnitine | 0.27 | 0.21 | 0.22 | 0.30 | 0.22 | 0.24 | 0.21 |
| 2H3-acetylcarnitine | 0.89 | 0.21 | 0.82 | 0.92 | 0.46 | 0.46 | 0.74 |
| 2H3-propionylcarnitine | 0.21 | 0.16 | 0.16 | 0.20 | 0.19 | 0.20 | 0.20 |
| 2H3-butyrylcarnitine | 3.39 | 0.63 | 1.34 | 1.53 | 0.77 | 0.92 | 1.08 |
| 2H9-isovalerylcarnitine | 0.20 | 0.13 | 0.15 | 0.17 | 0.19 | 0.20 | 0.19 |
| 2H3-octanoylcarnitine | 0.18 | 0.12 | 0.14 | 0.17 | 0.16 | 0.21 | 0.20 |
| 2H9-myristoylcarnitine | 0.20 | 0.14 | 0.14 | 0.17 | 0.20 | 0.22 | 0.20 |
| 2H3-palmitoylcarnitne | 0.19 | 0.16 | 0.15 | 0.18 | 0.23 | 0.23 | 0.21 |
| 5th percentile | 0.16 | 0.13 | 0.14 | 0.17 | 0.16 | 0.18 | 0.14 |
| Median | 0.20 | 0.16 | 0.16 | 0.20 | 0.21 | 0.21 | 0.20 |
| 95th percentile | 2.96 | 0.30 | 0.79 | 0.89 | 0.76 | 0.55 | 0.79 |
| Batch 1 | Batch 2 | Batch 3 | Batch 4 | Batch 5 | Batch 6 | Batch 7 | RSD | |
|---|---|---|---|---|---|---|---|---|
| Propionic aciduria | ||||||||
| Propionylcarnitine | 40.23 | 66.57 | 47.17 | 70.07 | 61.18 | 52.29 | 52.66 | 0.19 |
| Glycine | 12.99 | 20.75 | 17.28 | 16.42 | 23.48 | 24.54 | 10.12 | 0.30 |
| Propionylglycine | 7.89 | 7.69 | 9.26 | 7.54 | 12.69 | 9.09 | 6.10 | 0.24 |
| Lysinuric protein intolerance | ||||||||
| Citrulline | 23.86 | 26.55 | 18.98 | 29.23 | 24.10 | 30.03 | 22.51 | 0.15 |
| Glutamine | 3.32 | 3.40 | 3.56 | 4.68 | 3.39 | 4.81 | 2.07 | 0.26 |
| Lysine | −2.07 | −2.13 | −1.89 | −2.07 | −2.25 | −1.97 | −1.71 | 0.09 |
| Phenylketonuria | ||||||||
| Phenylalanine | 34.29 | 17.93 | 16.79 | 23.13 | 21.74 | 16.21 | 14.19 | 0.33 |
| DBS #1 | DBS #2 | Plasma #1 | Plasma #2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient Diagnosis | Metabolite * | Z-sc. | Rank | Correct Diagn. | Z-sc. | Rank | Correct Diagn. | Z-sc. | Rank | Correct Diagn. | Z-sc. | Rank | Correct Diagn. | |
| Urea cycle | OTC deficiency | Orotic acid | 5.7 | 1 | Yes (n = 2) | −0.5 | No (n = 3) | 11.7 | 2 | Yes (n = 2) | 2.3 | Yes (n = 1) | ||
| Uridine | 1.6 | −0.5 | 7.1 | 39.2 | 6 | |||||||||
| 5-Oxoproline | −0.7 | 0.2 | 0.4 | 9.0 | ||||||||||
| Uracil | −0.8 | −0.7 | 4.0 | 4.0 | ||||||||||
| Orotidine | 0.1 | −0.5 | −1.1 | 4.0 | ||||||||||
| L-Lysine | 0.0 | −0.2 | 0.3 | 3.3 | ||||||||||
| Citrulline | −0.3 | −1.8 | −7 | −0.6 | −2.8 | −7 | ||||||||
| Branched-chain amino acid metabolism | MSUD | Ketoleucine | 23.3 | 2 | Yes (n = 4) | 3.0 | 20 | Yes (n = 3) | 65.0 | 7 | Yes (n = 4) | 13.3 | 17 | Yes (n = 3) |
| (n = Iso) leucine | 12.4 | 6 | 0.4 | 37.4 | 10 | 24.7 | 7 | |||||||
| 2-Hydroxy-3-methylbutyr. acid | 9.4 | 8 | −0.4 | 579.1 | 1 | 234.8 | 1 | |||||||
| Alpha-ketoisovaleric acid | 4.8 | 2.2 | 39.5 | 9 | 21.2 | 9 | ||||||||
| IVA | Isovalerylcarnitine | 137.9 | 1 | Yes (n = 2) | 42.5 | 2 | Yes (n = 2) | 84.7 | 1 | Yes (n = 1) | 92.5 | 1 | Yes (n = 3) | |
| 3-Hydroxyisovaleric acid | 0.0 | −0.1 | 0.4 | 0.1 | ||||||||||
| 3-MCC | 3-Hydroxyisovaleric acid | 5.4 | 1 | Yes (n = 2) | 33.1 | 1 | Yes (n = 2) | 17.8 | 4 | No (n = 1) | 825.8 | 1 | Yes (n = 2) | |
| 3-Methylcrotonylglycine | 0.7 | 22.8 | 2 | −0.1 | 2.7 | |||||||||
| Isovalerylcarnitine | 0.6 | −1.2 | 7.0 | 11 | 0.7 | |||||||||
| 3-Hydroxyisovalerylcarnitine | 0.6 | −1.6 | −0.2 | 0.0 | ||||||||||
| MMA | Propionylcarnitine | 13.3 | 2 | Yes (n = 3) | 75.4 | 1 | Yes (n = 1) | |||||||
| Methylcitric acid | 7.3 | 4 | 4.3 | |||||||||||
| Methylmalonic acid | 0.2 | 16.6 | 4 | |||||||||||
| Methylmalonylcarnitine | 1.1 | 0.7 | ||||||||||||
| Lysine metabolism | GA-1 | Glutarylcarnitine | 18.6 | 2 | Yes (n = 4) | 4.9 | 10 | Yes (n = 2) | 26.3 | 1 | Yes (n = 3) | 27.9 | 5 | Yes (n = 3) |
| Glutaric acid | 7.9 | 3 | −0.9 | 6.4 | 17 | 71.6 | 3 | |||||||
| 3-hydroxyglutaric acid | −0.3 | 0.2 | 10.5 | 8 | 8.2 | 11 | ||||||||
| Glutaconic acid | −1.64 | 27.9 | 2 | |||||||||||
| Phenylalanine and tyrosine metabolism | PKU | Phenylalanine | 47.7 | 1 | Yes (n = 1) | 37.0 | 3 | Yes (n = 1) | ||||||
| Hydroxyphenylacetic acid | 10.9 | 4 | 1.9 | |||||||||||
| N-acetylphenylalanine | 6.3 | 9 | 7.0 | 22 | ||||||||||
| Tyrosine | −1.0 | −0.1 | ||||||||||||
| Tyrosinaemia | 4-Hydroxyphenyllactic acid | 150.7 | 1 | Yes (n = 1) | 125.6 | 2 | Yes (n = 1) | 206.5 | 1 | Yes (n = 3) | 263.5 | 13 | Yes (n = 3) | |
| Tyrosine | 26.2 | 3 | 15.6 | 6 | 35.0 | 3 | 33.7 | |||||||
| 4-Hydroxyphenylacetic acid | 4.6 | 6.3 | 9 | 2.2 | 2.0 | |||||||||
| 4-Hydroxyphenylpyruvic acid | 0.2 | 2.0 | 10.4 | 8 | 6.8 | |||||||||
| Succinylacetone | −1.5 | −1.2 | 0.2 | 1.1 | ||||||||||
| Sulphur amino acid metabolism | MAT1A deficiency | Methionine sulfoxide | 72.2 | 1 | Yes (n = 5) | 53.4 | 2 | Yes (n = 5) | 1106.7 | 1 | Yes (n = 1) | 632.2 | 1 | Yes (n = 3) |
| Methionine | 57.1 | 2 | 96.4 | 1 | 118.8 | 4 | 47.4 | 6 | ||||||
| S-adenosylmethionine | 0.5 | −0.3 | 0.1 | 0.3 | ||||||||||
| S-adenosylhomocysteine | −0.8 | 0.4 | 0.4 | 0.1 | ||||||||||
| CBS deficiency | Methionine sulfoxide | 22.4 | 2 | Yes (n = 4) | 778.9 | 1 | Yes (n = 2) | |||||||
| Methionine | 31.1 | 3 | 2.6 | |||||||||||
| Homocystine | 3.2 | 7 | 1.3 | |||||||||||
| Homocysteine | 2.6 | 12 | 2.2 | |||||||||||
| MTHFR deficiency | Homocysteine thiolactone | 28.0 | 1 | Yes (n = 6) | 7.5 | 3 | Yes | −0.3 | No (n = 1) | 0.1 | No (n = 3) | |||
| Homocystine | 1.1 | 4.8 | 9 | (n = 3) | −0.2 | 0.3 | ||||||||
| Methionine | 0.2 | 0.0 | −2.4 | −20 | −2.3 | −12 | ||||||||
| Molybdenum cofactor deficiency | Xanthine | 59.3 | 1 | Yes (n = 1) | 40.7 | 3 | Yes | 55.5 | 7 | Yes (n = 1) | ||||
| Alpha amino adipic semialdeh. | 3.4 | 1.5 | (n = 1) | 6.9 | ||||||||||
| Cysteine-S-sulfate | −0.9 | 0.6 | 11.8 | 22 | ||||||||||
| Cysteine | −1.0 | −2.6 | −2.1 | −14 | ||||||||||
| Uric acid | −1.4 | −0.8 | −2.6 | −5 | ||||||||||
| Serine and glycine metabolism | NKH | Glycine | 3.7 | 18 | Yes (n = 2) | 2.0 | No (n = 3) | 3.4 | Yes (n = 3) | 2.2 | No (n = 3) | |||
| 3-PGDH deficiency | Serine | 5.1 | 1 | No (n = 3) | 0.8 | No | −2.5 | −4 | Yes (n = 2) | −2.4 | −6 | Yes (n = 2) | ||
| Glycine | 2.1 | −0.1 | (n = 3) | −1.6 | −1.8 | |||||||||
| Proline metabolism | OAT deficiency | Proline | 4.0 | 11 | Yes (n = 6) | 4.0 | Yes (n = 5) | |||||||
| Ornithine | 2.8 | 18 | −0.8 | |||||||||||
| Amino acid transport | LPI | Citrulline | 8.5 | 2 | Yes (n = 3) | 16.1 | 13 | Yes (n = 3) | ||||||
| Serine | 6.2 | 3 | 2.4 | |||||||||||
| Proline | 6.4 | 4 | 0.2 | |||||||||||
| Threonine | 5.7 | 7 | 0.6 | |||||||||||
| Lysine | −2.0 | −7 | −1.3 | |||||||||||
| Ornithine | −1.5 | 0.6 | ||||||||||||
| Arginine | −1.0 | −1.0 | ||||||||||||
| Fatty acid oxidation | VLCAD deficiency | C14:1 carnitine | 28.9 | 1 | Yes (n = 1) | 0.6 | No (n = 3) | 7.3 | 34 | Yes (n = 1) | 5.8 | Yes (n = 1) | ||
| C14:2 carnitine | 15.7 | 2 | 1.4 | 7.6 | 33 | 2.8 | ||||||||
| C14-carnitine | 3.7 | 1.5 | 1.4 | 2.4 | ||||||||||
| LCHAD deficiency | C14-OH carnitine | 3.1 | 35 | Yes (n = 1) | 8.3 | 14 | Yes (n = 2) | 8.2 | Yes (n = 2) | |||||
| C16-OH carnitine | 3.0 | 37 | 22.7 | 2 | 37.3 | 12 | ||||||||
| C16-OH:1 carnitine | 1.5 | 23.8 | 1 | 41.6 | 11 | |||||||||
| C18-OH carnitine | 0.7 | 21.9 | 3 | 29.8 | 17 | |||||||||
| MCAD deficiency | C8-carnitine | 56.5 | 1 | Yes (n = 2) | 111.5 | 1 | Yes (n = 3) | 189.3 | 1 | Yes (n = 3) | 143.4 | 1 | Yes (n = 2) | |
| C6-carnitine | 7.3 | 6 | 16.0 | 3 | 51.7 | 2 | 55.7 | 2 | ||||||
| C10:1-carnitine | 1.7 | 8.1 | 7 | 24.9 | 4 | 11.6 | 5 | |||||||
| C10-carnitine | 1.1 | 2.6 | 7.3 | 12 | 3.2 | |||||||||
| OCTN2 deficiency | L-Carnitine | −2.0 | Yes (n = 1) | −1.3 | Yes (n = 4) | −2.4 | −3 | Yes (n = 2) | −2.3 | −6 | Yes (n = 1) | |||
| Acetylcarnitine | −1.9 | −0.9 | −2.5 | −1 | −2.5 | −9 | ||||||||
| C16-carnitine | −1.7 | −1.3 | −1.1 | −0.3 | ||||||||||
| C16:1-carnitineC18-carnitine | −2.6–1.7 | −5 | −1.1–1.7 | −2 | −1.3–0.6 | −1.8–0.9 | ||||||||
| C18:1-carnitine | −2.3 | −12 | −1.8 | −1 | −1.1 | −1.0 | ||||||||
| CPT1 deficiency | L-Carnitine | 19.0 | 1 | Yes (n = 2) | 19.0 | 1 | Yes (n = 6) | −2.7 | −84 | No (n = 2) | 1.8 | No (n = 2) | ||
| C0/(n = C16 + C18) ratio | 10.3 | 3 | 8.4 | 3 | −1.6 | −0.3 | ||||||||
| C16-carnitine | −3.1 | −1 | −1.8 | −5 | −2.7 | −82 | −0.2 | |||||||
| C18-carnitine | −2.6 | −3 | −2.2 | −2 | −1.1 | −0.6 | ||||||||
| C18:1-carnitine | −2.6 | −4 | −2.5 | −1 | 0.0 | 1.1 | ||||||||
| CPT2 deficiency | C16+C18:1/C2 ratio | 2.2 | 25 | Yes (n = 2) | 4.8 | 1 | Yes (n = 3) | −1.4 | Yes (n = 4) | 0.1 | Yes (n = 2) | |||
| Acetylcarnitine | −1.7 | −8 | −2.4 | −1 | 8.8 | 9 | 6.5 | 6 | ||||||
| C16-carnitine | −0.6 | −1.4 | 9.3 | 8 | 6.7 | 5 | ||||||||
| C18-carnitine | −0.6 | −1.7 | 4.1 | 3.1 | ||||||||||
| C18:1-carnitine | −0.7 | −1.8 | ||||||||||||
| Creatine biosynthesis | GAMT deficiency | Guanidoacetic acid | 20.9 | 1 | Yes (n = 2) | 39.2 | 2 | Yes (n = 1) | 25.1 | 1 | Yes (n = 3) | 35.9 | 1 | Yes (n = 1) |
| Creatine | −1.4 | −1.2 | 1.8 | −1.7 | ||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haijes, H.A.; Willemsen, M.; Van der Ham, M.; Gerrits, J.; Pras-Raves, M.L.; Prinsen, H.C.M.T.; Van Hasselt, P.M.; De Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites 2019, 9, 12. https://doi.org/10.3390/metabo9010012
Haijes HA, Willemsen M, Van der Ham M, Gerrits J, Pras-Raves ML, Prinsen HCMT, Van Hasselt PM, De Sain-van der Velden MGM, Verhoeven-Duif NM, Jans JJM. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites. 2019; 9(1):12. https://doi.org/10.3390/metabo9010012
Chicago/Turabian StyleHaijes, Hanneke A., Marcel Willemsen, Maria Van der Ham, Johan Gerrits, Mia L. Pras-Raves, Hubertus C. M. T. Prinsen, Peter M. Van Hasselt, Monique G. M. De Sain-van der Velden, Nanda M. Verhoeven-Duif, and Judith J. M. Jans. 2019. "Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma" Metabolites 9, no. 1: 12. https://doi.org/10.3390/metabo9010012
APA StyleHaijes, H. A., Willemsen, M., Van der Ham, M., Gerrits, J., Pras-Raves, M. L., Prinsen, H. C. M. T., Van Hasselt, P. M., De Sain-van der Velden, M. G. M., Verhoeven-Duif, N. M., & Jans, J. J. M. (2019). Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites, 9(1), 12. https://doi.org/10.3390/metabo9010012