An UPLC-MS/MS Assay to Measure Glutathione as Marker for Oxidative Stress in Cultured Cells

 ,
,
Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
2.2. Equipment
2.3. Cultured Primary Fibroblasts
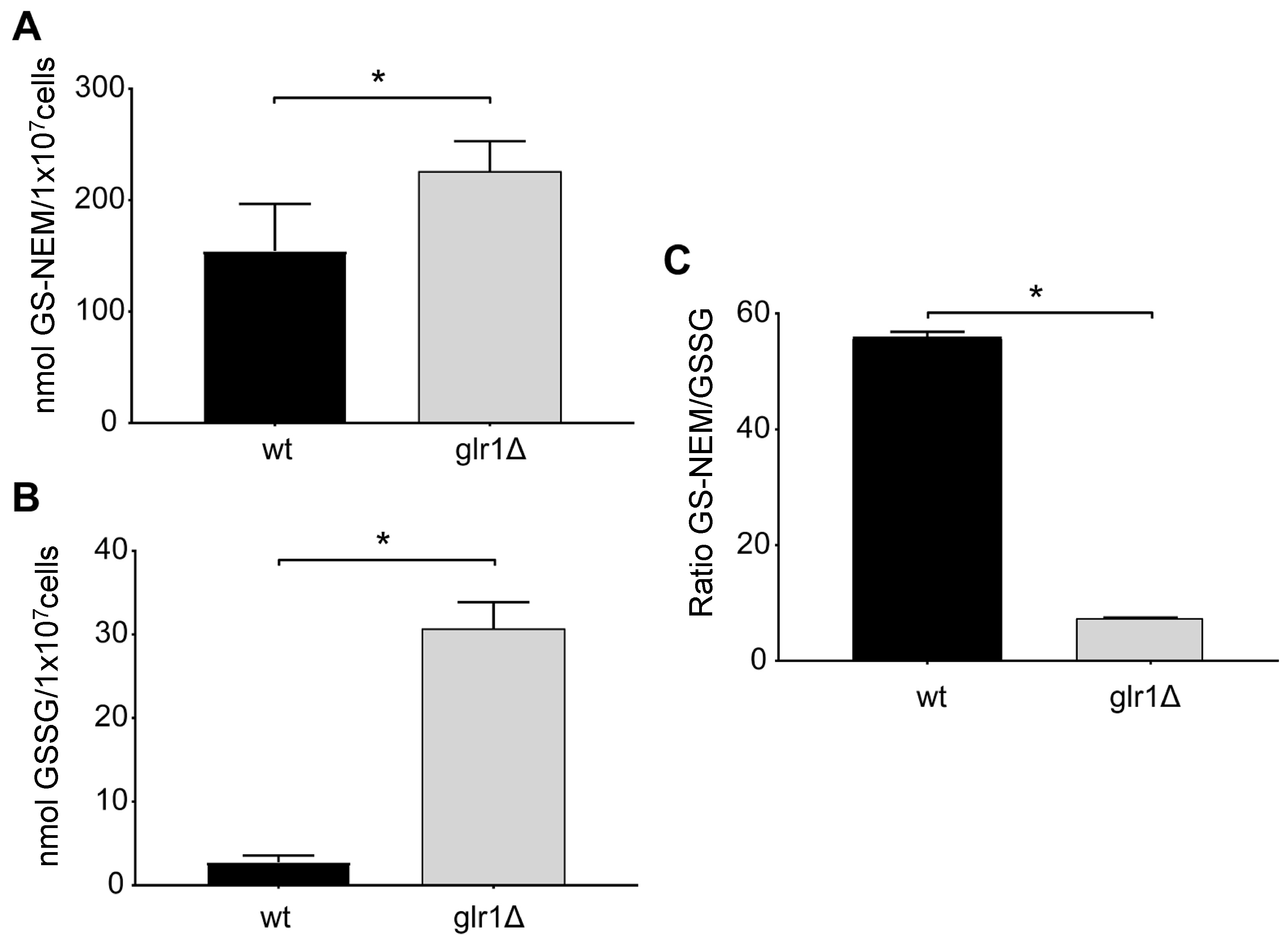
2.4. Saccharomyces Cerevisiae
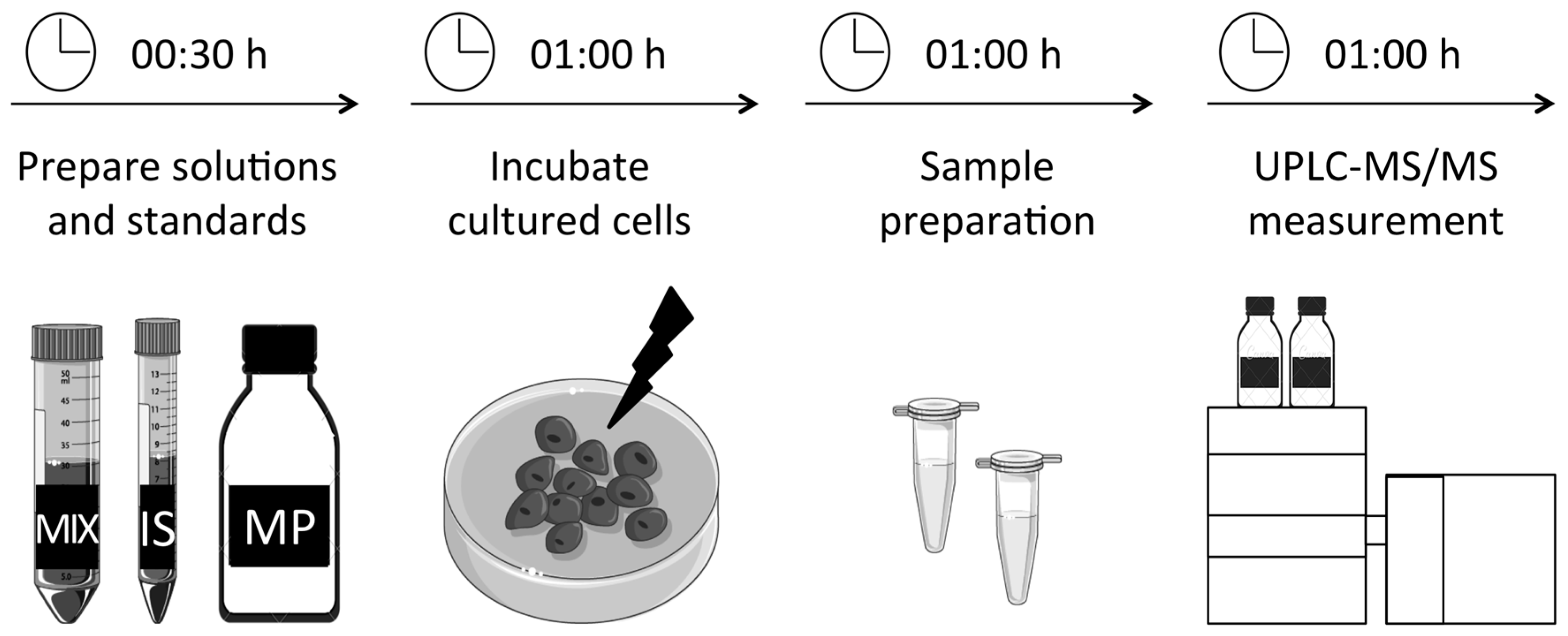
3. Procedure
3.1. Preparation of Solutions and Standards. Time for Completion: 00:30 Hours
- Prepare the NEM stock solution freshly on the day of experiment. Dissolve NEM in Milli-Q water to a final concentration of 20 mM.When using S. cerevisiae, prepare wash solutions containing NEM in Milli-Q water. Per sample, prepare 300 µL of wash solution containing 200 mM NEM, and 2 mL of wash solution containing 50 mM NEM.
- Prepare the precipitation solution freshly on the day of the experiment. Per sample, the precipitation solution consists of 280 µL NEM and 700 µL acetonitrile. Cool on ice prior to use.
- Prepare the internal standard mix. Per sample, the internal standard mix contains 10 µL labelled GSH (stock solution 650 µM, in Milli-Q) and 10 µL labelled GSSG (stock solution 3.25 µM, in Milli-Q) (see Section 5. Reagents Setup for details). Cool on ice prior to use.
- Prepare the mobile phases for the UPLC system. Mobile phase A consists of 0.1% formic acid in water, mobile phase B consists of 0.1% formic acid in acetonitrile.
3.2. Incubation and Preparation of Primary Fibroblasts. Time for Completion: 02:00 Hours
- Seed cells in 6-well plates the day prior to the experiment.Important: seed the cells in order to reach > 50% confluency on the day of the experiment.
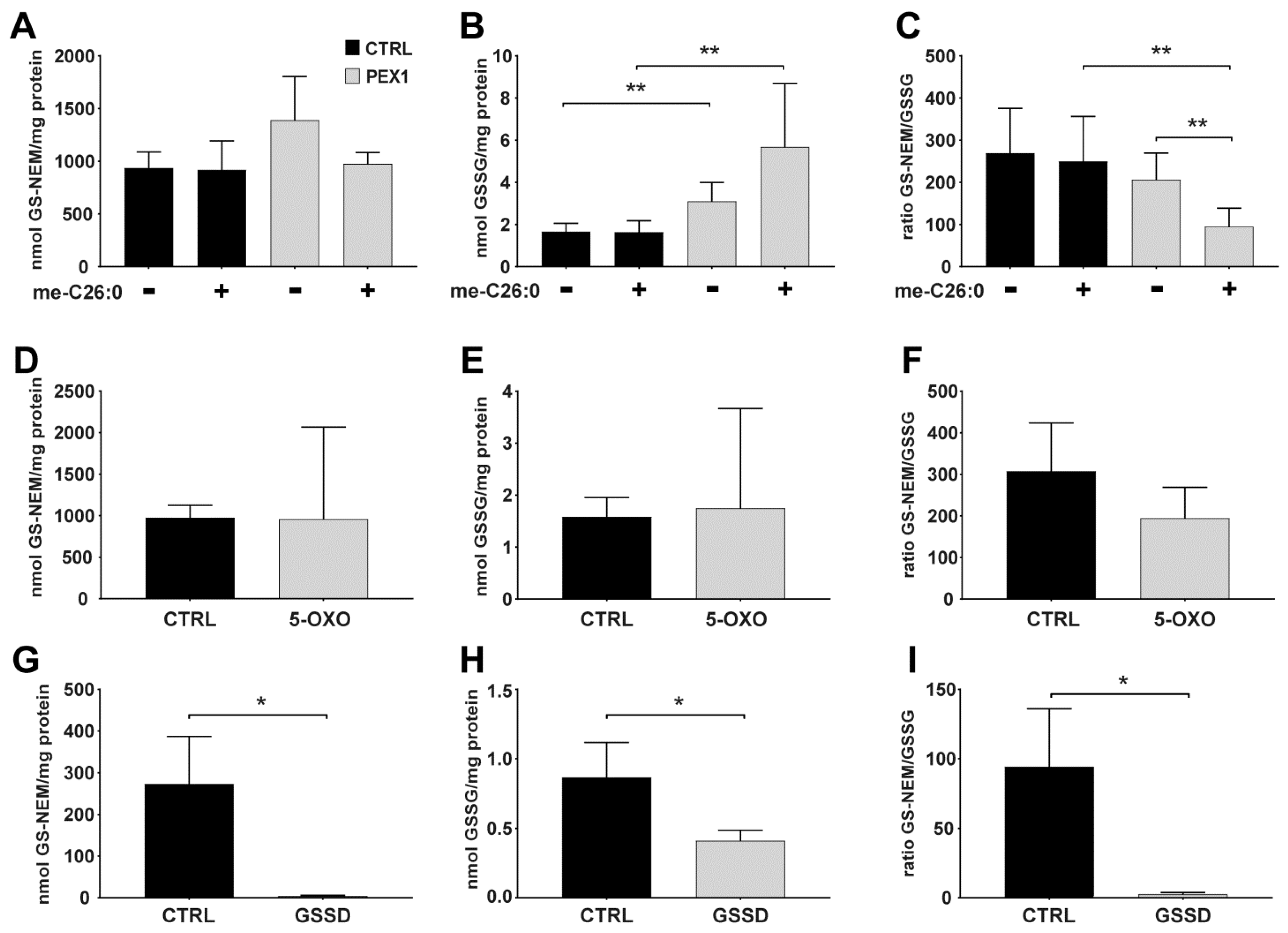
- OPTIONAL STEP On the next day, remove the culture medium of the cultured cells and add medium containing the chemicals of interest to the cells. For our experiments (see Figure 2 for details), we prepared stock solutions of BSO in Milli-Q water, stock solutions of menadione in ethanol, and stock solutions of me-C26:0 in DMSO. The methyl ester of C26:0 (me-C26:0) was prepared as described previously [7]. Incubate the cells for the desired time.Important: the maximum volume of added solvent may not exceed 1% v/v of the cell culture medium. As control, incubate cells with vehicle only, i.e., using an equal amount of solvent as was used for the incubations with the chemicals of interest.
- Take the cultured cells from the incubator and place the 6-well plate on ice.
- Remove the cell culture medium quickly and thoroughly, e.g., by suction.
![Metabolites 09 00045 i001]() CRITICAL STEP Add ice-cold precipitation solution and internal standard immediately to avoid auto-oxidation. Carefully shake the plate to make sure the entire surface of the well has been covered.
CRITICAL STEP Add ice-cold precipitation solution and internal standard immediately to avoid auto-oxidation. Carefully shake the plate to make sure the entire surface of the well has been covered.- Harvest the cells by scraping using a cell scraper.
- Collect both cells and supernatant in a 2.0 mL microcentrifuge tube.
- Centrifuge the samples at 5000× g for 10 min at 4 °C.
- Transfer the supernatant to a new microcentrifuge tube.
- Evaporate the supernatant under a nitrogen stream at 45 °C (duration ca. 00:30 hours).
- Re-suspend the pellet in 100 µL of mobile phase A.
![Metabolites 09 00045 i002]() PAUSE STEP The sample can be stored at −20 °C until analysis.
PAUSE STEP The sample can be stored at −20 °C until analysis.- Dry the cell pellet at room temperature.
- Re-suspend the cell pellet in 200 µL NaOH in Milli-Q water (0.2 M concentration).
![Metabolites 09 00045 i003]() PAUSE STEP The sample can be stored at −20 °C until analysis.
PAUSE STEP The sample can be stored at −20 °C until analysis.- Determine the protein concentration of the cell pellet, e.g., using the bicinchoninic acid assay [8].



3.3. Incubation and Preparation of S. cerevisae. Time for Completion: 02:00 Hours
- On the day prior to the experiment, shift the yeast cells from minimal ethanol growth medium (see Section 2.3 for details) to oleate/ethanol growth medium, consisting of 25 mM potassium phosphate (pH = 6.0), 3 g/L yeast extract, 5 g/L Peptone, 3.2 g/L oleate solution (12 g oleate + 20 mL tween-80), and 20 g/L ethanol.Important: allow the cells to grow in minimal medium for at least 24 h before switching to oleate/ethanol medium. Incubate for 17 h at 28 °C.
- Determine OD at 600 nm.
- Take 1 mL of yeast culture with OD = 1.0 and add 300 µL of wash solution (200 mM NEM).
- Mix briefly by vortexing or shaking.
- Centrifuge at 7000 rpm for 1 min at 4 °C.
- Remove the supernatant.
- Add 1 mL of wash solution (50 mM NEM) to the cell pellet.
- Vortex for 10 s.
- Centrifuge at 7000 rpm for 1 min at 4 °C.
- Repeat step 7–10 for a second wash.
- Remove the supernatant.
- Add ice-cold precipitation solution and internal standard.
- Add glass beads.
- Vortex for 5 min at 4 °C.
- Centrifuge at 7000 rpm for 1 min at 4 °C.
- Transfer the supernatant to a new microcentrifuge tube.
- Evaporate the supernatant under a nitrogen stream at 45 °C (duration ca. 00:30 hours).
- Re-suspend the pellet in 100 µL of mobile phase A.
![Metabolites 09 00045 i004]() PAUSE STEP The sample can be stored at −20 °C until analysis.
PAUSE STEP The sample can be stored at −20 °C until analysis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.4. UPLC-MS/MS Measurement. Time for Completion: 01:00 Hours
- Use 10 µL of sample to inject onto a UPLC HSS T3 1.8 µm column (2.1 × 100 mm) using a UPLC system.
- Set the column temperature to 50 °C and the flow rate to 400 µL/min.
- Use the following chromatographic conditions: 99.9% A to 2% B/98% A in 2 min., 2–3.5 min. to 30% B/70% A, to 95% B/5% A in 0.1 min., 3.51–4.5 min. isocratic 95% B/5% A, to 99.9% A in 0.1 min. The equilibration time with solvent A is 1.5 min.
- Use the triple-quadrupole mass spectrometer in positive electrospray ionization (ESI) mode.
- Use nitrogen as nebulizing gas and argon as collision gas at a pressure of 2.5 × 103 mbar.
- Set the capillary voltage to 3.5 kV and the source temperature to 120 °C. Set the desolvation gas flow to 900 L/h and the desolvation temperature to 350 °C.
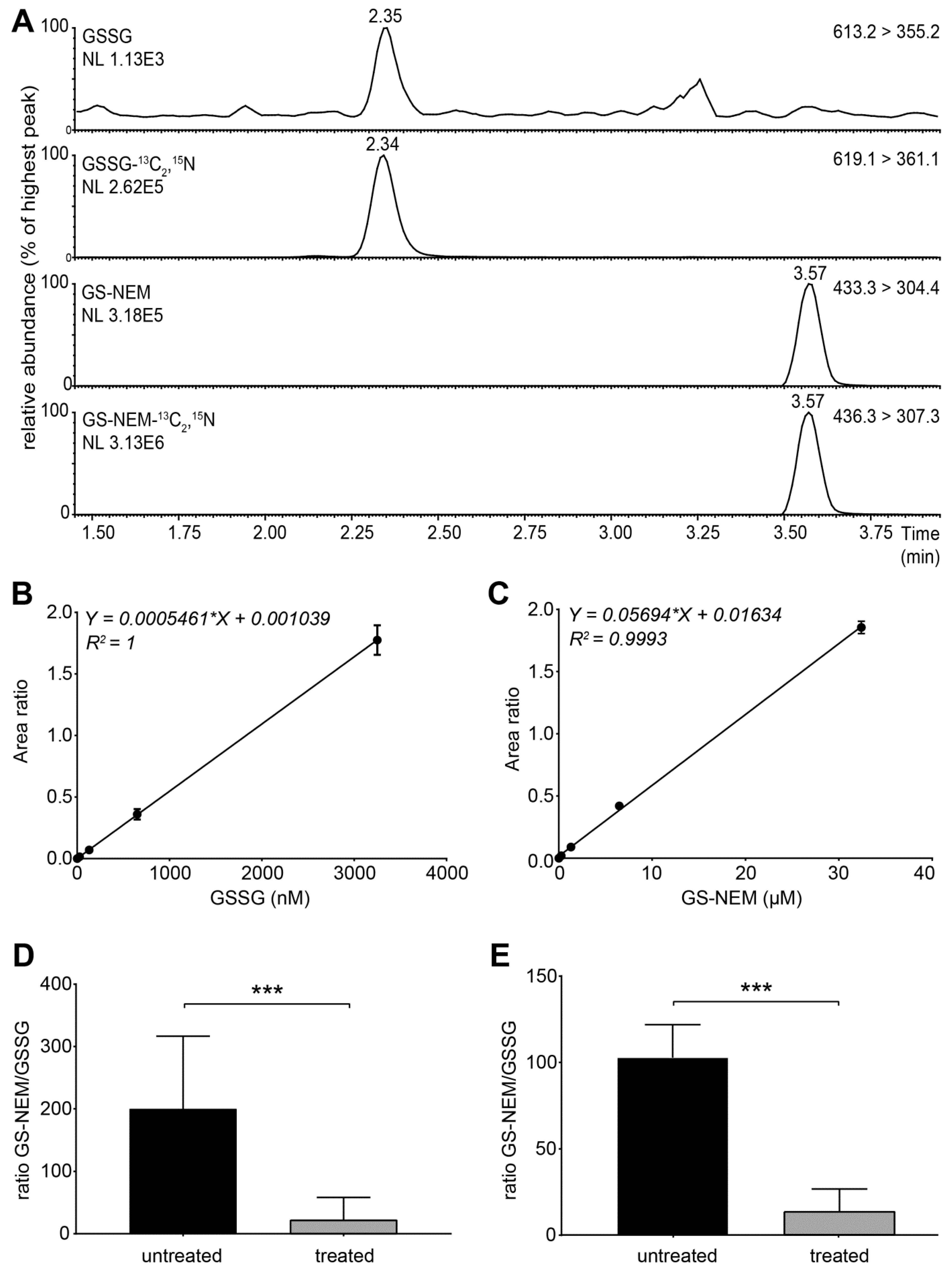
- Set the instrument to multiple reaction monitoring (MRM) in the positive ionization mode to detect GSH-NEM*, GSSG and the labelled internal standards. Use the following transitions for detection: m/z 311 > 165 (GSH-13C2,15N), m/z 308 >162 (GSH), m/z 433 > 304 (GS-NEM), m/z 436 > 307 (GS-NEM-13C2,15N), m/z 613 > 355 (GSSG)**, and m/z 619 > 361 (GSSG-13C4,15N2) (Figure S2). *NEM reacts with free sulfhydryl groups and thus forms a derivative with GSH molecules (GS-NEM). Accordingly, we detected the 13C, 15N-labelled internal standard for GSH as GS-NEM (Figure 2A). **For GSSG, we detected both singly and doubly charged molecular ions. The concentrations of GSSG calculated based on both ions were similar, but the formation of doubly charged GSSG ions was less reproducible in several experiments (data not shown). Therefore, we used the singly charged ion as precursor for quantification of GSSG (Figure 2A).
4. Results
4.1. Assay Validation and Data Analysis
4.2. Applying the Method to Cells Exposed to Exogenous and Endogenous Stress Inducers
4.3. Glutathione Levels in Cultured Cells with Different Genetic Defects in Glutathione Metabolism
4.4. Glutathione Levels in the Yeast S. cerevisiae
5. Reagents Setup
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B
References
- Iwasaki, Y.; Saito, Y.; Nakano, Y.; Mochizuki, K.; Sakata, O.; Ito, R.; Saito, K.; Nakazawa, H. Chromatographic and mass spectrometric analysis of glutathione in biological samples. J. Chromatogr. Anal. Technol. Biomed. Life Sci. 2009, 877, 3309–3317. [Google Scholar] [CrossRef] [PubMed]
- Monostori, P.; Wittmann, G.; Karg, E.; Túri, S. Determination of glutathione and glutathione disulfide in biological samples: An in-depth review. J. Chromatogr. Anal. Technol. Biomed. Life Sci. 2009, 877, 3331–3346. [Google Scholar] [CrossRef] [PubMed]
- Camera, E.; Picardo, M. Analytical methods to investigate glutathione and related compounds in biological and pathological processes. J. Chromatogr. Anal. Technol. Biomed. Life Sci. 2002, 781, 181–206. [Google Scholar] [CrossRef]
- Giustarini, D.; Galvagni, F.; Tesei, A.; Farolfi, A.; Zanoni, M.; Pignatta, S.; Milzani, A.; Marone, I.M.; Dalle-Donne, I.; Nassini, R.; et al. Glutathione, glutathione disulfide, and S-glutathionylated proteins in cell cultures. Free Radic. Biol. Med. 2015, 89, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Fahrenholz, T.; Wolle, M.M.; Kingston, H.M.S.; Faber, S.; Kern, J.C.; Pamuku, M.; Miller, L.; Chatragadda, H.; Kogelnik, A. Molecular speciated isotope dilution mass spectrometric methods for accurate, reproducible and direct quantification of reduced, oxidized and total glutathione in biological samples. Anal. Chem. 2015, 87, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Wach, A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 1996, 12, 259–265. [Google Scholar] [CrossRef]
- Van De Beek, M.-C.; Ofman, R.; Dijkstra, I.; Wijburg, F.; Engelen, M.; Wanders, R.; Kemp, S. Lipid-induced endoplasmic reticulum stress in X-linked adrenoleukodystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2255–2265. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Chiou, T.J.; Tzeng, W.F. The roles of glutathione and antioxidant enzymes in menadione-induced oxidative stress. Toxicology 2000, 154, 75–84. [Google Scholar] [CrossRef]
- Ristoff, E.; Larsson, A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2007, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.M.; Collinson, L.P.; Roe, J.-H.; Dawes, I.W. Yeast glutathione reductase is required for protection against oxidative stress and is a target gene for yAP-1 transcriptional regulation. Mol. Microbiol. 1996, 21, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Haberhauer-Troyer, C.; Delic, M.; Gasser, B.; Mattanovich, D.; Hann, S.; Koellensperger, G. Accurate quantification of the redox-sensitive GSH/GSSG ratios in the yeast Pichia pastoris by HILIC-MS/MS. Anal. Bioanal. Chem. 2013, 405, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Le, A.; Niemi, A.K.; Kwan, T.; Cusmano-Ozog, K.; Enns, G.M.; Cowan, T.M. A new LC-MS/MS method for the clinical determination of reduced and oxidized glutathione from whole blood. J. Chromatogr. Anal. Technol. Biomed. Life Sci. 2013, 929, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-G.; Yim, J.; Lim, Y.; Kim, J.-H. Validation of a liquid chromatography tandem mass spectrometry method to measure oxidized and reduced forms of glutathione in whole blood and verification in a mouse model as an indicator of oxidative stress. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1019, 45–50. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herzog, K.; IJlst, L.; van Cruchten, A.G.; van Roermund, C.W.T.; Kulik, W.; Wanders, R.J.A.; Waterham, H.R. An UPLC-MS/MS Assay to Measure Glutathione as Marker for Oxidative Stress in Cultured Cells. Metabolites 2019, 9, 45. https://doi.org/10.3390/metabo9030045
Herzog K, IJlst L, van Cruchten AG, van Roermund CWT, Kulik W, Wanders RJA, Waterham HR. An UPLC-MS/MS Assay to Measure Glutathione as Marker for Oxidative Stress in Cultured Cells. Metabolites. 2019; 9(3):45. https://doi.org/10.3390/metabo9030045
Chicago/Turabian StyleHerzog, Katharina, Lodewijk IJlst, Arno G. van Cruchten, Carlo W.T. van Roermund, Wim Kulik, Ronald J. A. Wanders, and Hans R. Waterham. 2019. "An UPLC-MS/MS Assay to Measure Glutathione as Marker for Oxidative Stress in Cultured Cells" Metabolites 9, no. 3: 45. https://doi.org/10.3390/metabo9030045
APA StyleHerzog, K., IJlst, L., van Cruchten, A. G., van Roermund, C. W. T., Kulik, W., Wanders, R. J. A., & Waterham, H. R. (2019). An UPLC-MS/MS Assay to Measure Glutathione as Marker for Oxidative Stress in Cultured Cells. Metabolites, 9(3), 45. https://doi.org/10.3390/metabo9030045