Fat is the most variable of the major components of ruminant milk, which can be affected by diet and the proportion of ruminal VFA [
7]. Previous studies have revealed that there was an association between shifts in the rumen VFA patterns and the decrease in yield of milk fat [
7,
8]. Coch et al. [
8] observed that diet-induced MFD cattle was related to the shifts in molar proportions of ruminal VFA. In consistency with these reports, in the present study, the cows in TR group encountered a decrease in percentage of acetate and butyrate and an increase in propionate percentage compared with CON group. Thus, the main reason for the MFD occurrence could be the decrease in percentage of butyrate and acetate.
Saliva production is an important characteristic of the ruminant digestive process because saliva acts as a buffering agent that helps to preserve optimum pH levels in the rumen. Saliva secretion occurs continuously but is more secreted during eating and ruminating [
9]. Rumination is a natural behavior of foregut fermenting species and it can stimulate the saliva secretion which maintains an optimum rumen environment. By decreasing the forage particle size through pelleting [
10], various studies have determined pelleted diets’ possessions on the feeding behavior of dairy cows. In this study, compared with TMR, pelleted diet altered the feeding behavior of dairy cows because of its own characteristics, and we found that the rumination time of dairy cows in TR group was decreased compared with CON group, and this was caused by short forage particle size of TR diet. TR diet had a higher proportion of maize with additional higher proportion of corn germ meal and corn husk, which provided large amounts of readily digestible carbohydrates. Mechanical activation and high temperature would change the physical structure of the starch and could enhance the starch degradability in the rumen [
11]. In the present study, the ingredients of the TR diet were chopped, ground and pelleted through high exit temperature (84 °C) by the pelleted machine. This result indicated that TR diet could enhance the degradability of starch in the rumen and caused MFD in dairy cows. Expectedly, cows in the TR group had a lower time of rumination, which may cause the lower ruminal pH which was similar to the previous studies [
12,
13]. These results indicated that the time of rumination in dairy cows can partially change the ruminal pH by means of saliva secretion.
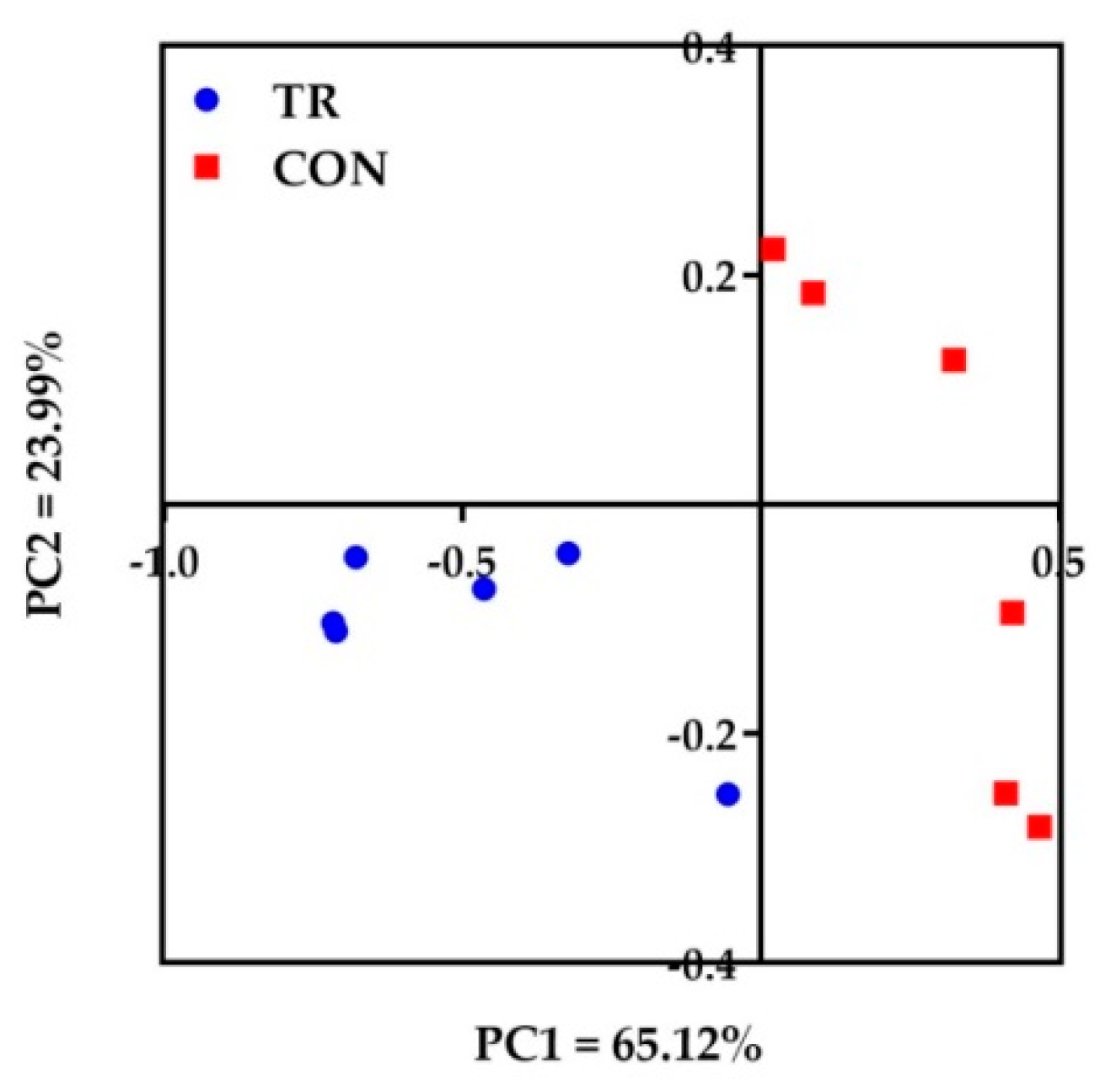
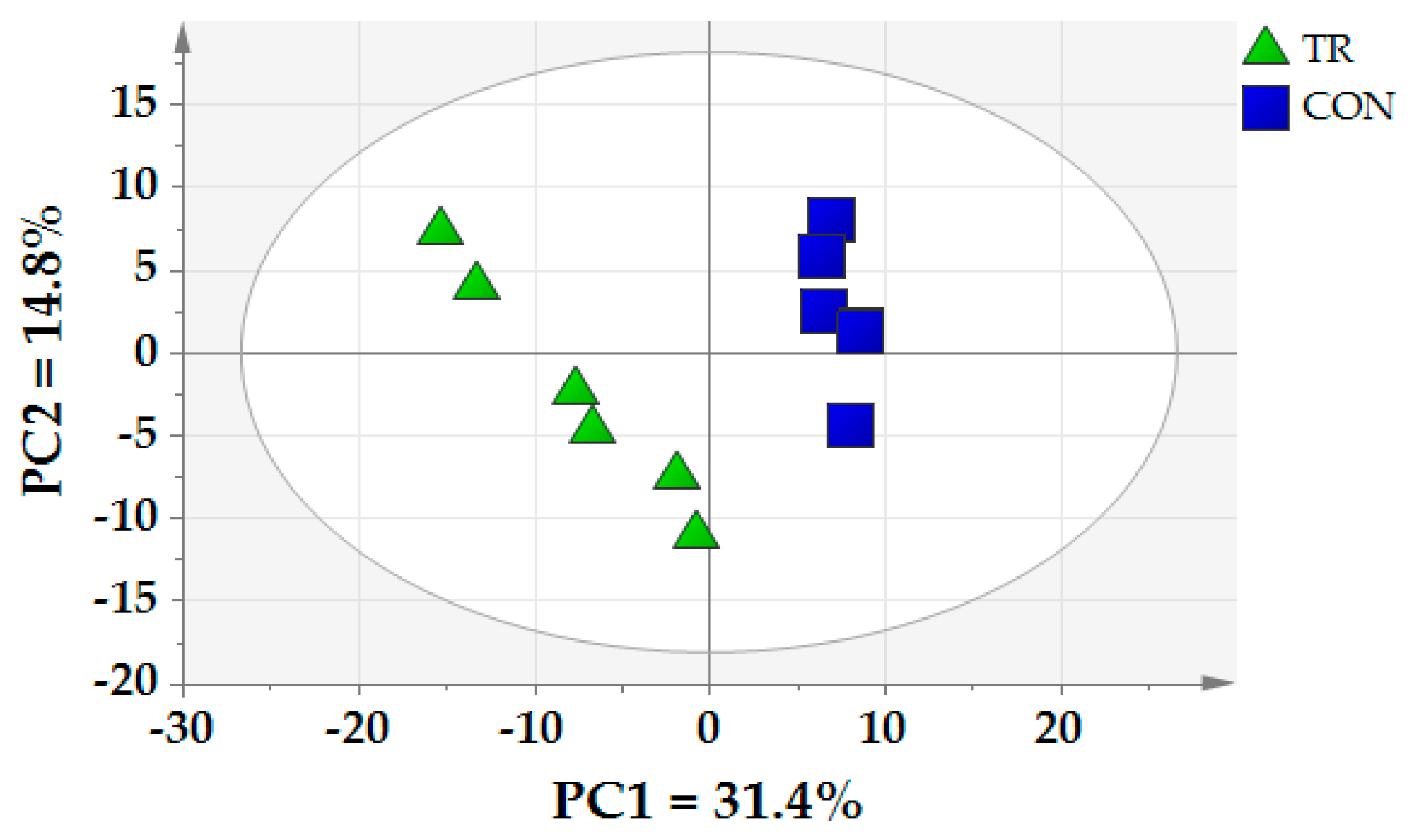
The richness and diversity of ruminal bacterial microbiota are important indicators of their normal physiological state. In the present study, the lower Chao 1 and Shannon index in TR group suggested that the microbial community composition was altered and tended to be less diverse of dairy cows in TR group. Belanche et al. [
14] found that compared with fiber rich diet, starch rich diet can decrease the bacterial and fungal diversity in the rumen. The reason was certain microorganisms were sensitive to nitrogen and rumen ammonia concentration. The accumulation of amines could exert negative effect on the ruminal microbiota. In our study, the metabolites of certain amines were increased in TR group which was consistent with Belanche et al. [
14]. We also found that the relative abundance of Bacteroidetes was higher and the Firmicutes and Proteobacteria were lower in TR group in the present study. A replacement of Firmicutes and Proteobacteria by Bacteroidetes was associated with the transition from the forage to concentrate diets [
15]. This indicated the Bacteroidetes became the dominant bacteria by the high starch diet. Our results in the present study confirmed the previous reports that
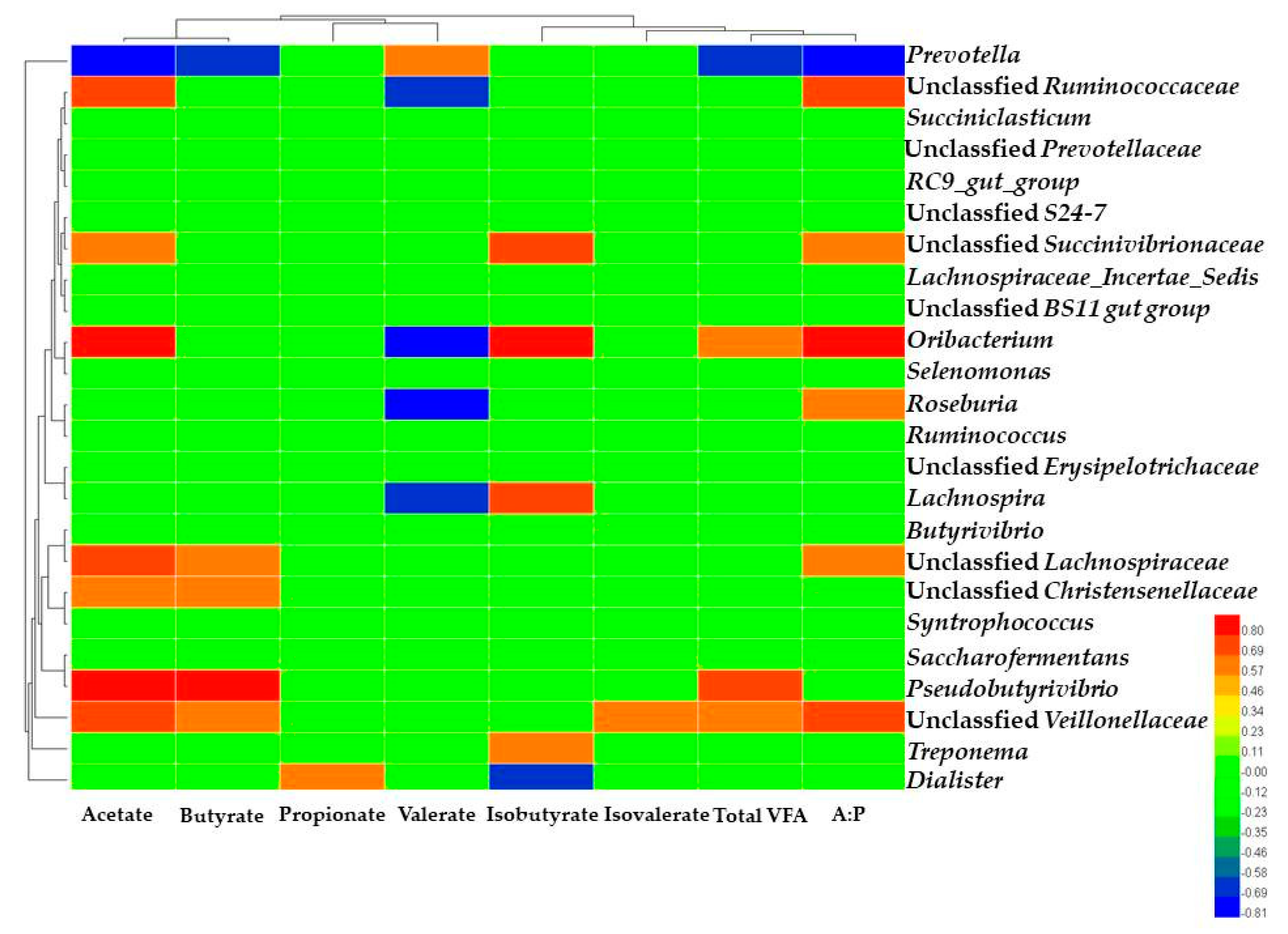
Prevotella was the most abundant genus of adult dairy cows [
16]. Reduced eating time with increased starch content in diets could result in the high relative abundance of
Prevotella, which could be explained by the increased proportion of ruminal fermentable substrates and the reduction in ruminal pH [
17]. Correlation analysis revealed the relative abundance of
Prevotella was negatively correlated with the acetate, butyrate concentration and the ratio of acetate to propionate in the present study, which indicated that high relative abundance of
Prevotella changed the ruminal fermentation type. The main reason was that TR diet provided larger proportion of ruminal fermentable substrates, changed the ruminal pH and caused the shift of the ruminal bacterial community composition, which altered the VFA production. In this study, high-starch diet provided large amounts of readily digestible carbohydrates which could increase the relative abundance of
Prevotella, and the
Prevotella taxa would participate in the carbohydrates digesting process. Meanwhile, the lower ruminal pH could provide the suitable conditions for
Prevotella to survive, which was consistent with James [
18]. Consistent with the findings by Liu et al. [
19], some higher abundance of unclassified groups, containing unclassified
Lachnospiraceae and unclassified
Ruminococcaceae were observed in rumen of dairy cattle. Kim et al. [
20] found that these two unclassified bacterial groups might play a significant role in fiber digestion of rumen. In the present study, the dairy cows in TR group had lower relative abundance of unclassified
Lachnospiraceae and unclassified
Ruminococcaceae, correlation analysis revealed that unclassified
Lachnospiraceae and unclassified
Ruminococcaceae had positive correlation with the acetate concentration and the ratio of acetate to propionate, which indicated that both of the bacteria could be intensively concerned with high starch diets fermentation in the rumen. The ruminal microbiome provides many physiological functions that are required by the host dairy cow. To define the potential functions of the microbiota in ruminal fluid samples, the PICRUSt was used to infer putative metagenomes from the 16S rRNA gene profiles [
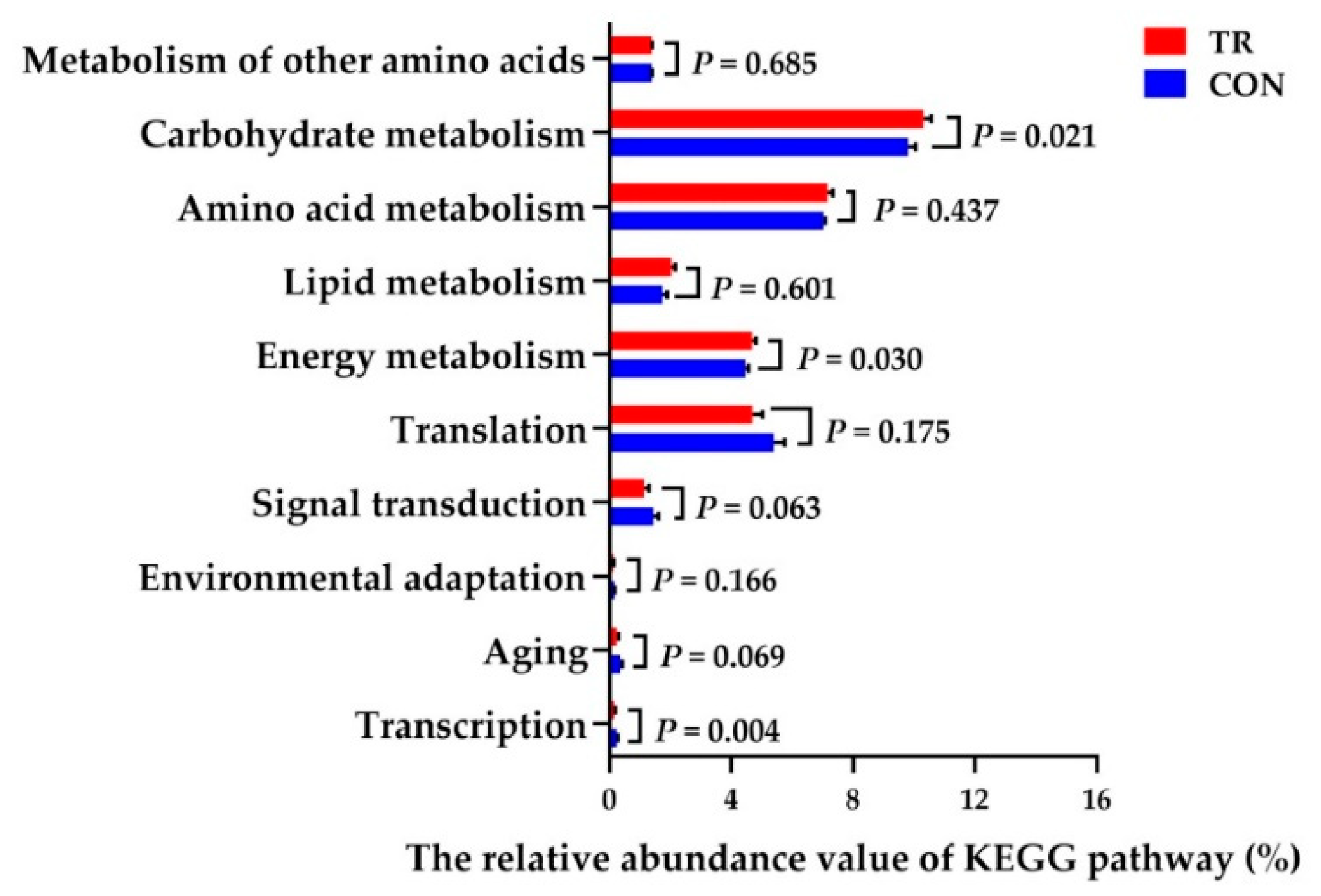
21]. Results showed that the ruminal microbiota had an enhanced capacity to affect carbohydrate metabolism and energy metabolism, while it had a decreased capacity of transcription and aging of dairy cows in TR group. These results implied that rumen bacteria and its functions could respond quickly to high levels of starch in TR diets, and adapt to the diets by enhancing the carbohydrate and energy metabolism.
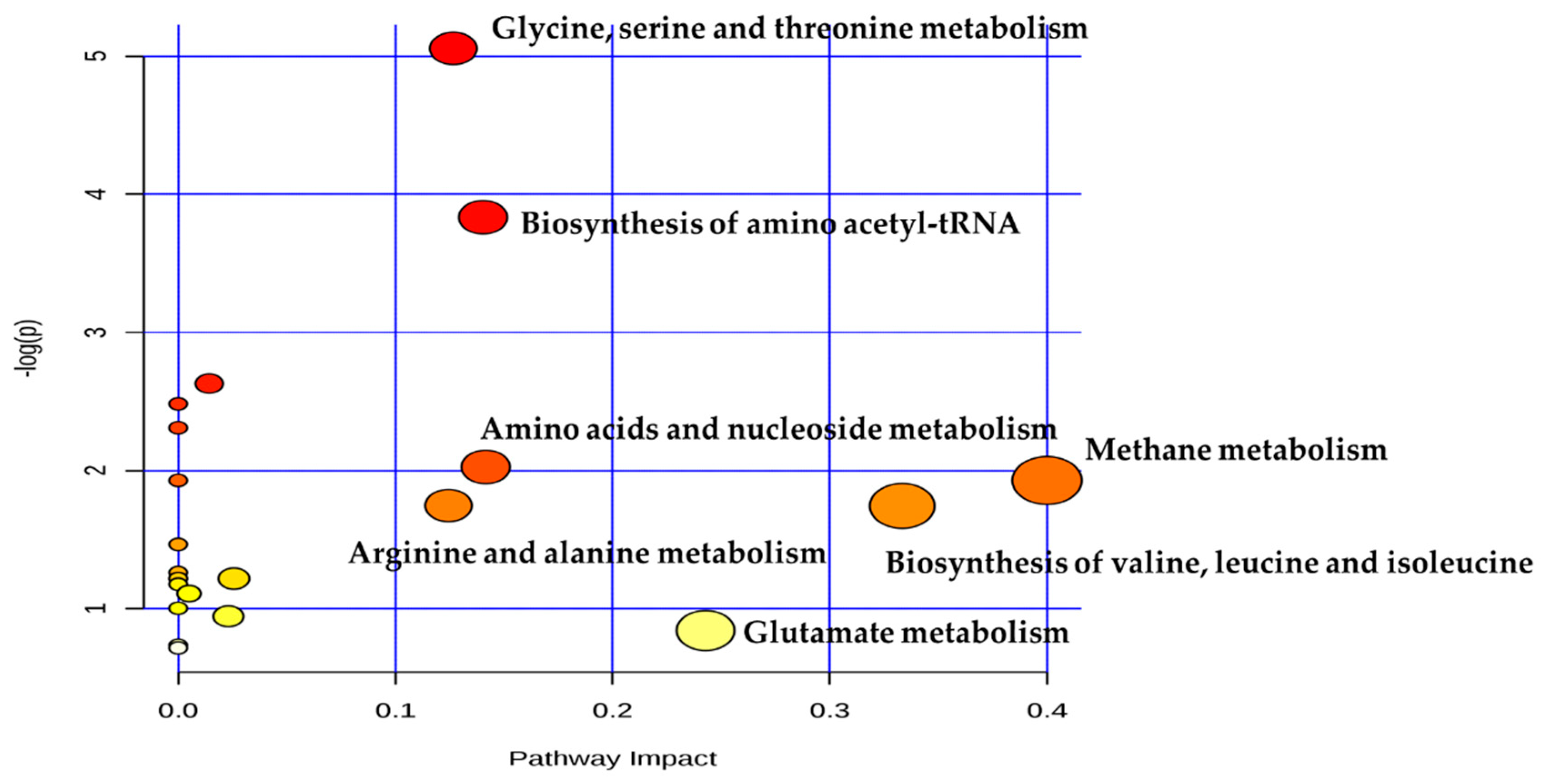
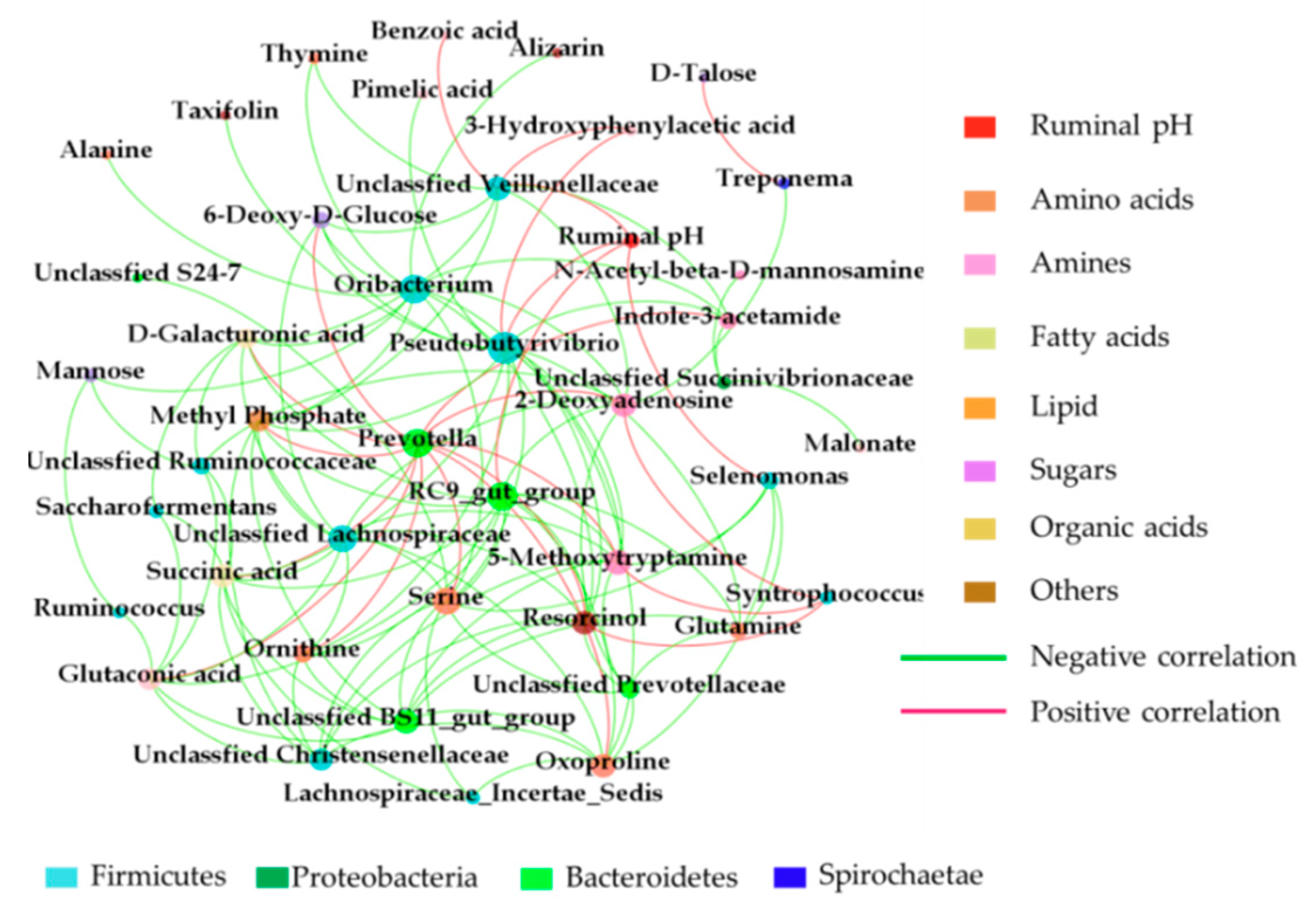
Metabolomics analysis revealed that the levels of glutamine, isoleucine, ornithine, oxyproline, alanine and serine were higher in dairy cows of TR group. Trent et al. [
22] found that the concentration of alanine was concerned with the demise of Gram-negative and Gram-positive bacteria. In this study, correlation analysis revealed that alanine was negatively correlated with
Oribacterium. Therefore, the increased concentration of alanine in the rumen of dairy cows with MFD appeared to be caused by the alternation in the abundance of
Oribacterium in this study. From the catabolic pathway of arginine in many microorganisms [
23], ornithine has been revealed to be formed by ornithine carbamoyl-transferase. Correlation analysis revealed that the ornithine had high positive correlation with the
Prevotella and negative correlation with the
Pseudobutyrivibrio. In this study, the metabolism of ornithine was higher in TR group. This finding indicated that the genus of
Prevotella and
Pseudobutyrivibrio may be correlated with the ornithine production through the catabolic pathway of arginine in dairy cows with MFD. From the present study, the levels of biogenic amines including
n-acetyl-beta-
d-mannosamine, malonamide, 5-methoxytryptamine, indole-3-acetamide were increased in TR group. Ruminants possibly obtained biogenic amines from both diets and microbial metabolites in the rumen and the biogenic amines were mainly instigated from the decarboxylation of convinced amino acids [
24]. Moreover, the enriched pathways including biosynthesis of valine, leucine, isoleucine and amino-acetyl-tRNA biosynthesis was also observed in cows of TR group. These findings indicated that ruminal microbial bacteria enhanced amino acids metabolism. The possible explanation for the increase in the amino acid metabolism is supported by the lower ruminal pH, and previous studies revealed that the increased activity of bacterial amino acid decarboxylases was owing to the decreased ruminal pH [
25,
26].
For dicarboxylic acids, malonate, pimelic acid, azelaic acid and glutaconic acid were identified in this study. The dicarboxylic acids were produced by the biohydrogenation of the non-conjugated linoleic acids formed from linoleic [
27,
28]. Previous studies suggested that the rate of bio-hydrogenation in the rumen was linked to the milk fat depression (MFD) [
29]. Correlation analysis revealed that the glutaconic acid had a higher negative correlation with unclassified
Ruminococcaceae, unclassified
Lachnospiraceae and unclassified
Christensenellaceae, meanwhile, the benzoic acid and 3-hydroxyphenylacetic acid had a higher positive correlation with the unclassified
Veillonellaceae. These results indicated that these unclassified bacteria such as unclassified
Ruminococcaceae, unclassified
Lachnospiraceae, unclassified
Christensenellaceae and unclassified
Veillonellaceae may be related with the fatty acids metabolism combined with the rate of bio-hydrogenation. This finding was consistent with Huws [
30] who observed that unclassified
Clostridiales and
Ruminococcaceae exert significant effect on bio-hydrogenation. However, these data did not conclude that these bacteria are definitively elaborate in bio-hydrogenation. Further studies should aim to investigate the exact functions of these bacteria in bio-hydrogenation.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}